Evaluating KOMPUTE - Body Composition Data
Coby Warkentin and Donghyung Lee
2023-07-03
- Import necessary packages
- Preparing control phenotype
data
- Importing Body Composition Control Phenotype Dataset
- Visualizing measured phenotypes via a heatmap
- Exclude phenotypes with fewer than 15,000 observations
- Remove samples with fewer than 7 measured phenotypes
- Heapmap of filtered phenotypes
- Reforatting the dataset (long to wide)
- Visualizing phenotype distributions
- Rank Z transformation
- Conducting Principal Variance Component Analysis (PVCA)
- Batch effect removal using ComBat
- PVCA on ComBat residuals
- Supplementary figure
- Computing phenotypic correlations
- Preparation of IMPC
summary statistics data
- Loading Body Composition summary stat (IMPCv16)
- Visualizing gene-phenotype pair duplicates
- Consolidating muliple z-scores of a gene-phenotype pair using Stouffer’s Method
- Generating Z-score matrix (reformatting)
- Visualization of Phenotype-Gene Coverage
- Distribution of Z-Scores Across Phenotypes
- Estimation of Genetic Correlation Matrix Using Z-Scores
- Comparison of Phenotypic Correlation and Genetic Correlation Among Phenotypes
- Correlation Analysis Between Genetic Correlation Matrices Using Mantel’s Test
- Evaluating the KOMPUTE Imputation Algorithm
Last updated: 2023-07-03
Checks: 7 0
Knit directory: komputeExamples/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230110) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 0bd39c6. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: code/.DS_Store
Untracked files:
Untracked: analysis/kompute_test_app_v16.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/kompute_test_BC_v16.Rmd)
and HTML (docs/kompute_test_BC_v16.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 0bd39c6 | statsleelab | 2023-07-03 | typo fix |
| html | b343ff3 | statsleelab | 2023-07-03 | svd mc added |
| Rmd | dbdfbca | statsleelab | 2023-07-03 | svd mc added |
| Rmd | 16fe14d | statsleelab | 2023-07-03 | updated |
| Rmd | e938a74 | statsleelab | 2023-07-03 | updated |
| Rmd | 7ea04d7 | statsleelab | 2023-07-03 | svd mc added |
| html | 0ad952c | statsleelab | 2023-06-23 | kompute function input format changed |
| Rmd | fbd0f5c | statsleelab | 2023-06-23 | kompute input format changed |
| html | e7a365f | statsleelab | 2023-06-21 | pheno.cor added |
| Rmd | fd859e3 | statsleelab | 2023-06-21 | BC pheno cor added |
| html | 9b680ec | statsleelab | 2023-06-20 | doc updated |
| Rmd | d343f6a | statsleelab | 2023-06-20 | doc updated |
| html | d56cc7a | statsleelab | 2023-06-19 | Build site. |
| html | 989feb6 | statsleelab | 2023-01-10 | v16 update |
| Rmd | f4e1ab9 | statsleelab | 2023-01-10 | v16 update |
| html | 7685a09 | statsleelab | 2023-01-10 | first commit |
| Rmd | 32dd775 | statsleelab | 2023-01-10 | first commit |
Import necessary packages
rm(list=ls())
knitr::opts_chunk$set(message = FALSE, warning = FALSE)
library(data.table)
library(dplyr)
library(reshape2)
library(ggplot2)
library(tidyr) #spread
library(RColorBrewer)
library(circlize)
library(ComplexHeatmap)
library(patchwork)
library(ggpubr)Preparing control phenotype data
Importing Body Composition Control Phenotype Dataset
BC.data <- readRDS("data/BC.data.rds")
#dim(BC.data)Visualizing measured phenotypes via a heatmap
The heatmap below presents a visualization of the phenotypic measurements taken for each control mouse. The columns represent individual mice, while the rows correspond to the distinct phenotypes measured.
mtest <- table(BC.data$proc_param_name_stable_id, BC.data$biological_sample_id)
mtest <-as.data.frame.matrix(mtest)
#dim(mtest)
if(FALSE){
nmax <-max(mtest)
library(circlize)
col_fun = colorRamp2(c(0, nmax), c("white", "red"))
col_fun(seq(0, nmax))
ht = Heatmap(as.matrix(mtest), cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, col = col_fun,
row_names_gp = gpar(fontsize = 8), name="Count")
draw(ht)
}Exclude phenotypes with fewer than 15,000 observations
To maintain data quality and robustness, we will discard any phenotypes that have fewer than 15,000 recorded observations.
mtest <- table(BC.data$proc_param_name, BC.data$biological_sample_id)
#dim(mtest)
#head(mtest[,1:10])
mtest0 <- mtest>0
#head(mtest0[,1:10])
#rowSums(mtest0)
rmv.pheno.list <- rownames(mtest)[rowSums(mtest0)<15000]
#rmv.pheno.list
#dim(BC.data)
BC.data <- BC.data %>% filter(!(proc_param_name %in% rmv.pheno.list))
#dim(BC.data)
# number of phenotypes left
#length(unique(BC.data$proc_param_name))Remove samples with fewer than 7 measured phenotypes
mtest <- table(BC.data$proc_param_name, BC.data$biological_sample_id)
#dim(mtest)
#head(mtest[,1:10])
mtest0 <- mtest>0
#head(mtest0[,1:10])
#summary(colSums(mtest0))
rmv.sample.list <- colnames(mtest)[colSums(mtest0)<7]
#length(rmv.sample.list)
#dim(BC.data)
BC.data <- BC.data %>% filter(!(biological_sample_id %in% rmv.sample.list))
#dim(BC.data)
# number of observations to use
#length(unique(BC.data$biological_sample_id))Heapmap of filtered phenotypes
if(FALSE){
mtest <- table(BC.data$proc_param_name, BC.data$biological_sample_id)
dim(mtest)
mtest <-as.data.frame.matrix(mtest)
nmax <-max(mtest)
library(circlize)
col_fun = colorRamp2(c(0, nmax), c("white", "red"))
col_fun(seq(0, nmax))
pdf("~/Google Drive Miami/Miami_IMPC/output/measured_phenotypes_controls_after_filtering_BC.pdf", width = 10, height = 3)
ht = Heatmap(as.matrix(mtest), cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, col = col_fun,
row_names_gp = gpar(fontsize = 7), name="Count")
draw(ht)
dev.off()
}Reforatting the dataset (long to wide)
We restructure our data from a long format to a wide one for further processing and analysis.
BC.mat <- BC.data %>%
dplyr::select(biological_sample_id, proc_param_name, data_point, sex, phenotyping_center, strain_name) %>%
##consider weight or age in weeks
arrange(biological_sample_id) %>%
distinct(biological_sample_id, proc_param_name, .keep_all=TRUE) %>% ## remove duplicates, maybe mean() is better.
spread(proc_param_name, data_point) %>%
tibble::column_to_rownames(var="biological_sample_id")
head(BC.mat) sex phenotyping_center strain_name BC_BMC/Body weight BC_Body length
39638 female MRC Harwell C57BL/6NTac 0.0567631 NA
39639 female HMGU C57BL/6NCrl 0.0224897 NA
39640 female HMGU C57BL/6NTac 0.0214276 NA
39641 male HMGU C57BL/6NCrl 0.0191929 9.7
39642 female MRC Harwell C57BL/6NTac 0.0242145 NA
39643 female HMGU C57BL/6NCrl 0.0224004 9.4
BC_Body weight BC_Bone Area BC_Bone Mineral Content (excluding skull)
39638 22.12 13.51560 1.25560
39639 23.30 8.11161 0.52401
39640 23.20 6.85683 0.49712
39641 29.70 8.61072 0.57003
39642 25.27 8.42837 0.61190
39643 24.30 8.42616 0.54433
BC_Bone Mineral Density (excluding skull) BC_Fat mass BC_Fat/Body weight
39638 0.0929 6.9362 0.313571
39639 0.0646 6.1000 0.261803
39640 0.0725 3.3000 0.142241
39641 0.0662 7.1000 0.239057
39642 0.0726 3.4382 0.136059
39643 0.0646 6.9000 0.283951
BC_Lean mass BC_Lean/Body weight
39638 11.2569 0.508901
39639 14.7000 0.630901
39640 17.1000 0.737069
39641 21.7000 0.730640
39642 20.5392 0.812790
39643 15.3000 0.629630#dim(BC.mat)
#summary(colSums(is.na(BC.mat[,-1:-3])))Visualizing phenotype distributions
ggplot(melt(BC.mat), aes(x=value)) +
geom_histogram() +
facet_wrap(~variable, scales="free", ncol=5)+
theme(strip.text.x = element_text(size = 6))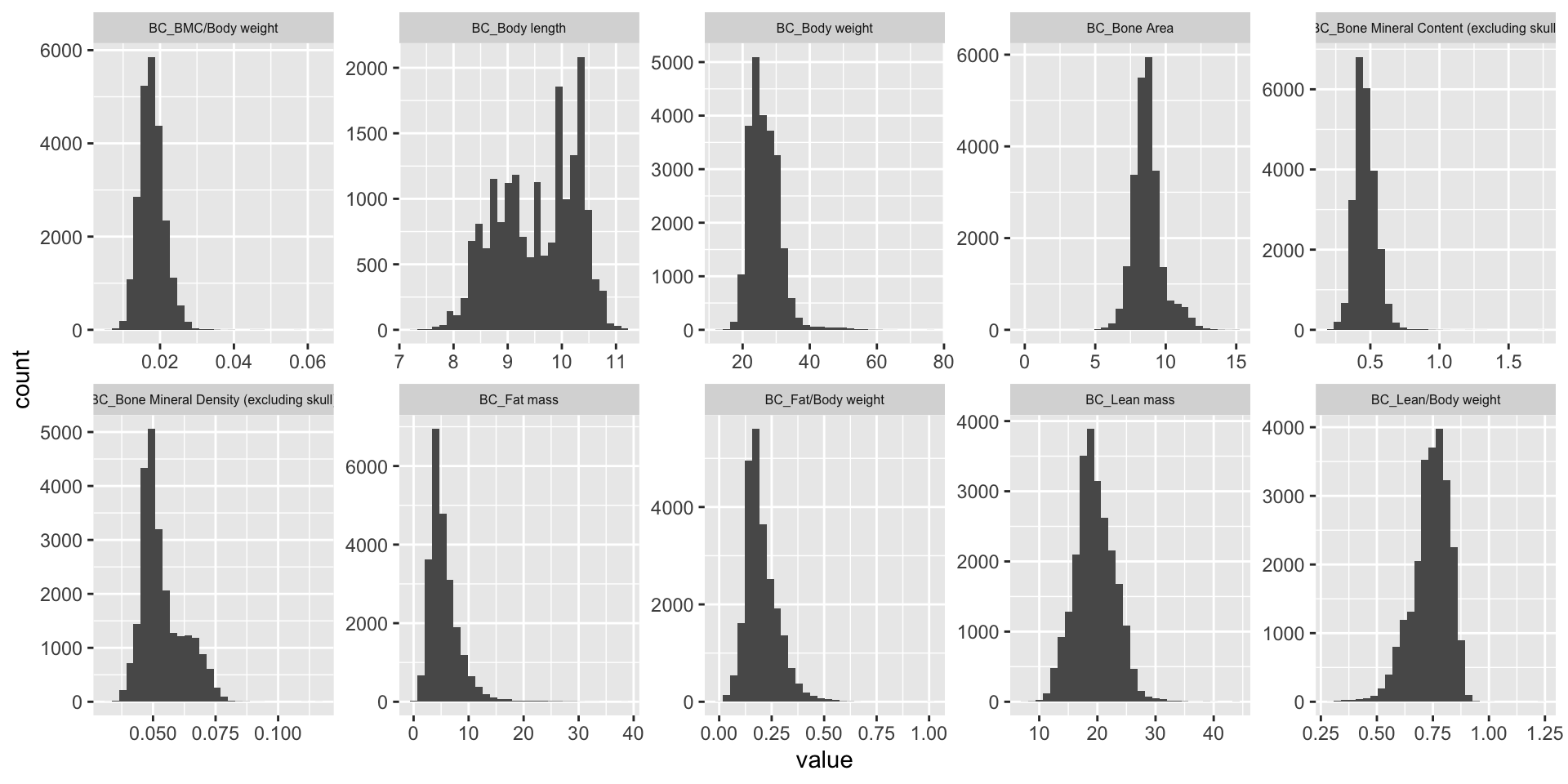
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Rank Z transformation
In this step, we conduct a rank Z transformation on the phenotype data to ensure that the data is normally distributed
library(RNOmni)
BC.mat.rank <- BC.mat
#dim(BC.mat.rank)
BC.mat.rank <- BC.mat.rank[complete.cases(BC.mat.rank),]
#dim(BC.mat.rank)
#dim(BC.mat)
BC.mat <- BC.mat[complete.cases(BC.mat),]
#dim(BC.mat)
BC.mat.rank <- cbind(BC.mat.rank[,1:3], apply(BC.mat.rank[,-1:-3], 2, RankNorm))
ggplot(melt(BC.mat.rank), aes(x=value)) +
geom_histogram() +
facet_wrap(~variable, scales="free", ncol=5)+
theme(strip.text.x = element_text(size = 6))![]()
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Conducting Principal Variance Component Analysis (PVCA)
In this step, we apply Principal Variance Component Analysis (PVCA) on the phenotype matrix data. PVCA is an approach that combines Principal Component Analysis (PCA) and Variance Component Analysis to quantify the proportion of total variance in the data attributed to each important covariate, in this case ‘sex’ and ‘phenotyping_center’.
First, we prepare our metadata which includes our chosen covariates. Any character variables in the metadata are then converted to factors. To avoid potential confounding, we check for associations between our covariates and drop ‘strain_name’ due to its strong association with ‘phenotyping_center’.
Next, we run PVCA on randomly chosen subsets of our phenotype data (for computational efficiency). Finally, we compute the average effect size across all random samples and visualize the results in a PVCA plot.
source("code/PVCA.R")
meta <- BC.mat.rank[,1:3] ## examining covariates sex, phenotyping_center, and strain_name
#head(meta)
#dim(meta)
#summary(meta) # variables are currently characters
meta[sapply(meta, is.character)] <- lapply(meta[sapply(meta, is.character)], as.factor)
#summary(meta) # now all variables are converted to factors
chisq.test(meta[,1],meta[,2])
Pearson's Chi-squared test
data: meta[, 1] and meta[, 2]
X-squared = 13.572, df = 10, p-value = 0.1934chisq.test(meta[,2],meta[,3])
Pearson's Chi-squared test
data: meta[, 2] and meta[, 3]
X-squared = 59770, df = 50, p-value < 2.2e-16meta<-meta[,-3] # phenotyping_center and strain_name strongly associated which could cause confounding in the PVCA analysis, so we drop 'strain_name'.
G <- t(BC.mat.rank[,-1:-3]) ## preparing the phenotype matrix data
set.seed(09302021)
# Perform PVCA for 10 random samples of size 1000 (more computationally efficient)
pvca.res <- matrix(nrow=10, ncol=3)
for (i in 1:10){
sample <- sample(1:ncol(G), 1000, replace=FALSE)
pvca.res[i,] <- PVCA(G[,sample], meta[sample,], threshold=0.6, inter=FALSE)
}
# Compute average effect size across the 10 random samples
pvca.means <- colMeans(pvca.res)
names(pvca.means) <- c(colnames(meta), "resid")
# Create PVCA plot
pvca.plot <- PlotPVCA(pvca.means, "PVCA of Phenotype Matrix Data")
pvca.plot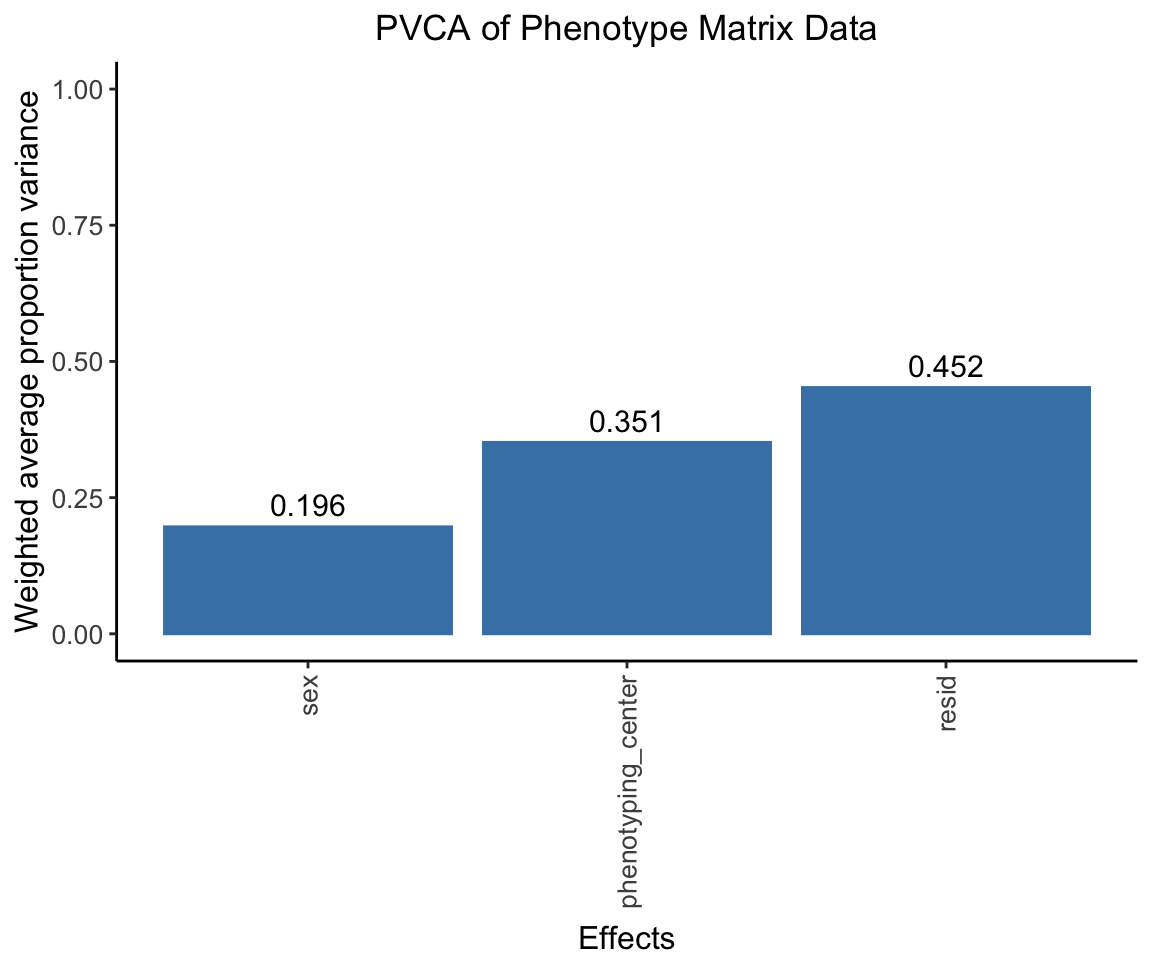
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
png(file="docs/figure/figures.Rmd/pvca_BC_1_v16.png", width=600, height=350)
pvca.plot
dev.off()quartz_off_screen
2 Batch effect removal using ComBat
We remove batch effects (the center effect) in the phenotype data set by using the ComBat method.
library(sva)
combat_komp = ComBat(dat=G, batch=meta$phenotyping_center, par.prior=TRUE, prior.plots=TRUE, mod=NULL)
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
#combat_komp[1:5,1:5]
#G[1:5,1:5] # for comparison, combat_komp is same form and same dimensions as GPVCA on ComBat residuals
After using ComBat to account for batch effects, we perform a PVCA on the residuals. We expect to observe a significantly reduced effect from the phenotyping centers.
set.seed(09302021)
# Perform PVCA for 10 random samples (more computationally efficient)
pvca.res.nobatch <- matrix(nrow=10, ncol=3)
for (i in 1:10){
sample <- sample(1:ncol(combat_komp), 1000, replace=FALSE)
pvca.res.nobatch[i,] <- PVCA(combat_komp[,sample], meta[sample,], threshold=0.6, inter=FALSE)
}
# Compute average effect size across samples
pvca.means.nobatch <- colMeans(pvca.res.nobatch)
names(pvca.means.nobatch) <- c(colnames(meta), "resid")
# Generate PVCA plot
pvca.plot.nobatch <- PlotPVCA(pvca.means.nobatch, "PVCA of Phenotype Matrix Data with Reduced Batch Effect")
pvca.plot.nobatch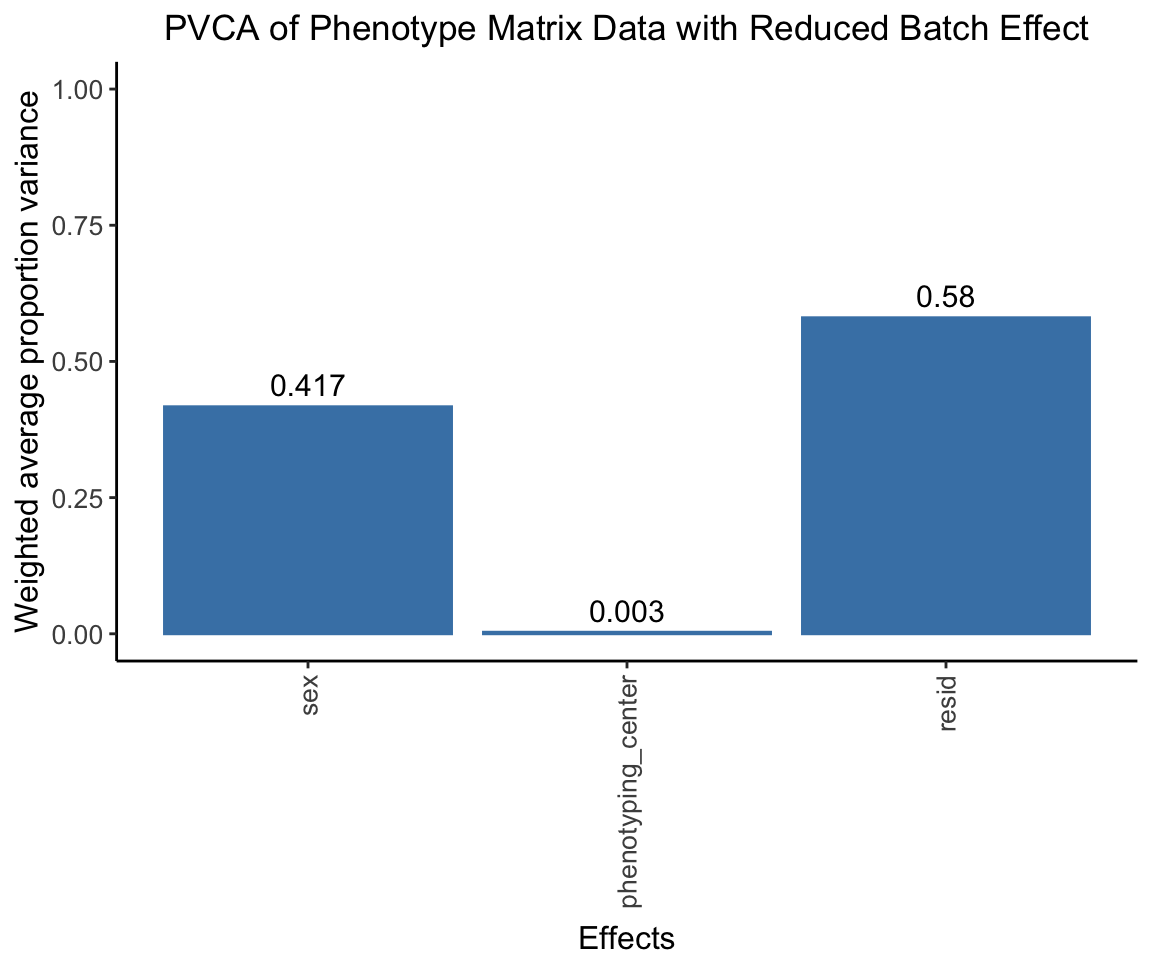
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
png(file="docs/figure/figures.Rmd/pvca_BC_2_v16.png", width=600, height=350)
pvca.plot.nobatch
dev.off()quartz_off_screen
2 Supplementary figure
# Supplementary Figure 1
pvca.plot <- pvca.plot +
labs(title="") +
scale_x_discrete(limits=names(pvca.means), labels=c("Sex", "Pheno center", "Residual")) +
theme(axis.text.x = element_text(angle=0, vjust=1, hjust=.5))
pvca.plot.nobatch <- pvca.plot.nobatch +
labs(y="", title="") +
scale_x_discrete(limits=names(pvca.means.nobatch), labels=c("Sex", "Pheno center", "Residual")) +
theme(axis.text.x = element_text(angle=0, vjust=1, hjust=.5))
pvca.plots <- ggarrange(pvca.plot, pvca.plot.nobatch, labels = "AUTO")
png(file="docs/figure/figures.Rmd/pvca_BC_supp_v16.png", width=700, height=400)
pvca.plots
dev.off()quartz_off_screen
2 Computing phenotypic correlations
We compute the phenotype correlations using different methods and compare them.
BC.cor.rank <- cor(BC.mat.rank[,-1:-3], use="pairwise.complete.obs") # pearson correlation coefficient
BC.cor <- cor(BC.mat[,-1:-3], use="pairwise.complete.obs", method="spearman") # spearman
BC.cor.combat <- cor(t(combat_komp), use="pairwise.complete.obs")
pheno.list <- rownames(BC.cor)
ht1 = Heatmap(BC.cor, show_column_names = F, row_names_gp = gpar(fontsize = 9), name="Spearm. Corr.")
draw(ht1)
ht2 = Heatmap(BC.cor.rank, show_column_names = F, row_names_gp = gpar(fontsize = 9), name="Corr. RankZ")
draw(ht2)
ht3 = Heatmap(BC.cor.combat, show_column_names = F, row_names_gp = gpar(fontsize = 9), name="Corr. ComBat")
draw(ht3)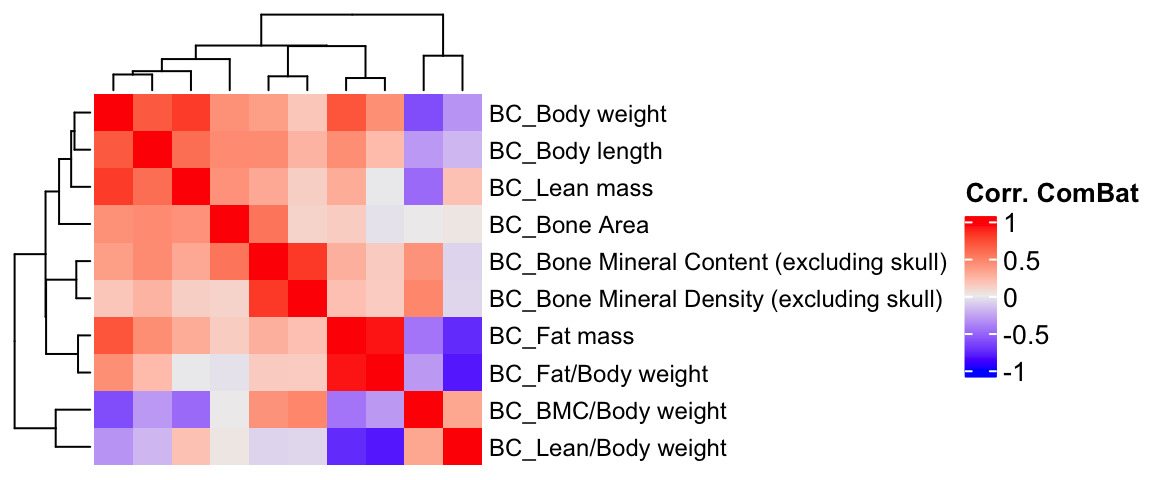
Preparation of IMPC summary statistics data
Loading Body Composition summary stat (IMPCv16)
BC.stat <- readRDS("data/BC.stat.v16.rds")
#dim(BC.stat)
table(BC.stat$parameter_name, BC.stat$procedure_name)
BC
BMC/Body weight 6943
Body length 6121
Bone Area 6945
Bone Mineral Content (excluding skull) 5825
Bone Mineral Density (excluding skull) 6945
Fat mass 2884
Fat/Body weight 7026
Lean mass 7028
Lean/Body weight 7026#length(unique(BC.stat$marker_symbol)) #6145
#length(unique(BC.stat$allele_symbol)) #6313
#length(unique(BC.stat$proc_param_name)) #9, number of phenotypes in association statistics data set
#length(unique(BC.data$proc_param_name)) #10, number of phenotypes in final control data
pheno.list.stat <- unique(BC.stat$proc_param_name)
pheno.list.ctrl <- unique(BC.data$proc_param_name)
#sum(pheno.list.stat %in% pheno.list.ctrl)
#sum(pheno.list.ctrl %in% pheno.list.stat)
# Identifying common phenotypes between statistics and control data
common.pheno.list <- sort(intersect(pheno.list.ctrl, pheno.list.stat))
common.pheno.list[1] "BC_BMC/Body weight"
[2] "BC_Body length"
[3] "BC_Bone Area"
[4] "BC_Bone Mineral Content (excluding skull)"
[5] "BC_Bone Mineral Density (excluding skull)"
[6] "BC_Fat mass"
[7] "BC_Fat/Body weight"
[8] "BC_Lean mass"
[9] "BC_Lean/Body weight" #length(common.pheno.list) # 9 - each data set had one phenotype not present in the other
# Filtering summary statistics to contain only common phenotypes
#dim(BC.stat)
BC.stat <- BC.stat %>% filter(proc_param_name %in% common.pheno.list)
#dim(BC.stat)
#length(unique(BC.stat$proc_param_name))
# Exclude 'Fat mass' due to limited gene availability
# only 2884 genes available in Fat mass
BC.stat <- BC.stat %>% filter(parameter_name != "Fat mass")
#dim(BC.stat)
#length(unique(BC.stat$proc_param_name))
table(BC.stat$parameter_name, BC.stat$procedure_name)
BC
BMC/Body weight 6943
Body length 6121
Bone Area 6945
Bone Mineral Content (excluding skull) 5825
Bone Mineral Density (excluding skull) 6945
Fat/Body weight 7026
Lean mass 7028
Lean/Body weight 7026common.pheno.list <- common.pheno.list[-6]
common.pheno.list[1] "BC_BMC/Body weight"
[2] "BC_Body length"
[3] "BC_Bone Area"
[4] "BC_Bone Mineral Content (excluding skull)"
[5] "BC_Bone Mineral Density (excluding skull)"
[6] "BC_Fat/Body weight"
[7] "BC_Lean mass"
[8] "BC_Lean/Body weight" Visualizing gene-phenotype pair duplicates
mtest <- table(BC.stat$proc_param_name, BC.stat$marker_symbol)
mtest <-as.data.frame.matrix(mtest)
nmax <-max(mtest)
col_fun = colorRamp2(c(0, nmax), c("white", "red"))
#col_fun(seq(0, nmax))
ht = Heatmap(as.matrix(mtest), cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, col = col_fun,
row_names_gp = gpar(fontsize = 8), name="Count")
draw(ht)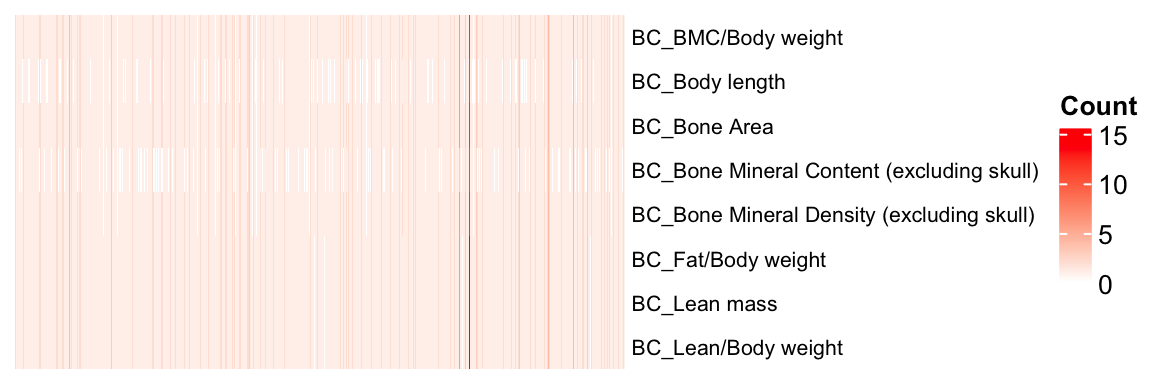
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Consolidating muliple z-scores of a gene-phenotype pair using Stouffer’s Method
## sum(z-score)/sqrt(# of zscore)
sumz <- function(z){ sum(z)/sqrt(length(z)) }
BC.z = BC.stat %>%
dplyr::select(marker_symbol, proc_param_name, z_score) %>%
na.omit() %>%
group_by(marker_symbol, proc_param_name) %>%
summarize(zscore = sumz(z_score)) ## combine z-scores
#dim(BC.z)Generating Z-score matrix (reformatting)
# Function to convert NaN to NA
nan2na <- function(df){
out <- data.frame(sapply(df, function(x) ifelse(is.nan(x), NA, x)))
colnames(out) <- colnames(df)
out
}
# Converting the long format of z-scores to a wide format matrix
BC.zmat = dcast(BC.z, marker_symbol ~ proc_param_name, value.var = "zscore",
fun.aggregate = mean) %>% tibble::column_to_rownames(var="marker_symbol")
BC.zmat = nan2na(BC.zmat) #convert nan to na
#dim(BC.zmat)Visualization of Phenotype-Gene Coverage
The heatmap illustrates tested (red) and untested (white) gene-phenotype pairs.
# Generate a matrix indicating where z-scores are present
id.mat <- 1*(!is.na(BC.zmat)) # multiply 1 to make this matrix numeric
#nrow(as.data.frame(colSums(id.mat)))
#dim(id.mat)
ht = Heatmap(t(id.mat),
cluster_rows = T, clustering_distance_rows ="binary",
cluster_columns = T, clustering_distance_columns = "binary",
show_row_dend = F, show_column_dend = F, # do not show dendrogram
show_column_names = F, col = c("white","red"),
row_names_gp = gpar(fontsize = 10), name="Missing")
draw(ht)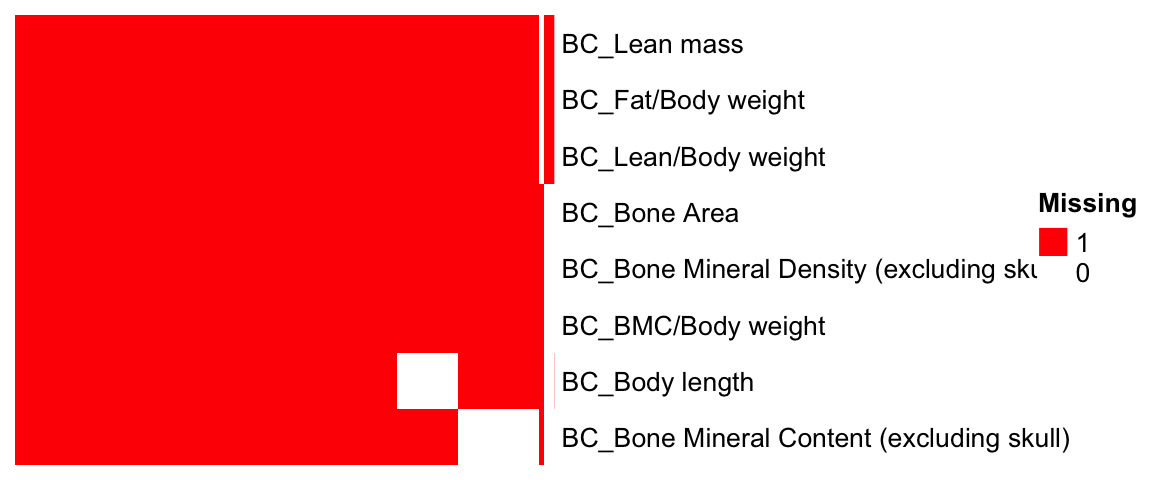
Distribution of Z-Scores Across Phenotypes
The histogram presents the distribution of association Z-scores for each phenotype.
ggplot(melt(BC.zmat), aes(x=value)) +
geom_histogram() +
facet_wrap(~variable, scales="free", ncol=5)+
theme(strip.text.x = element_text(size = 6))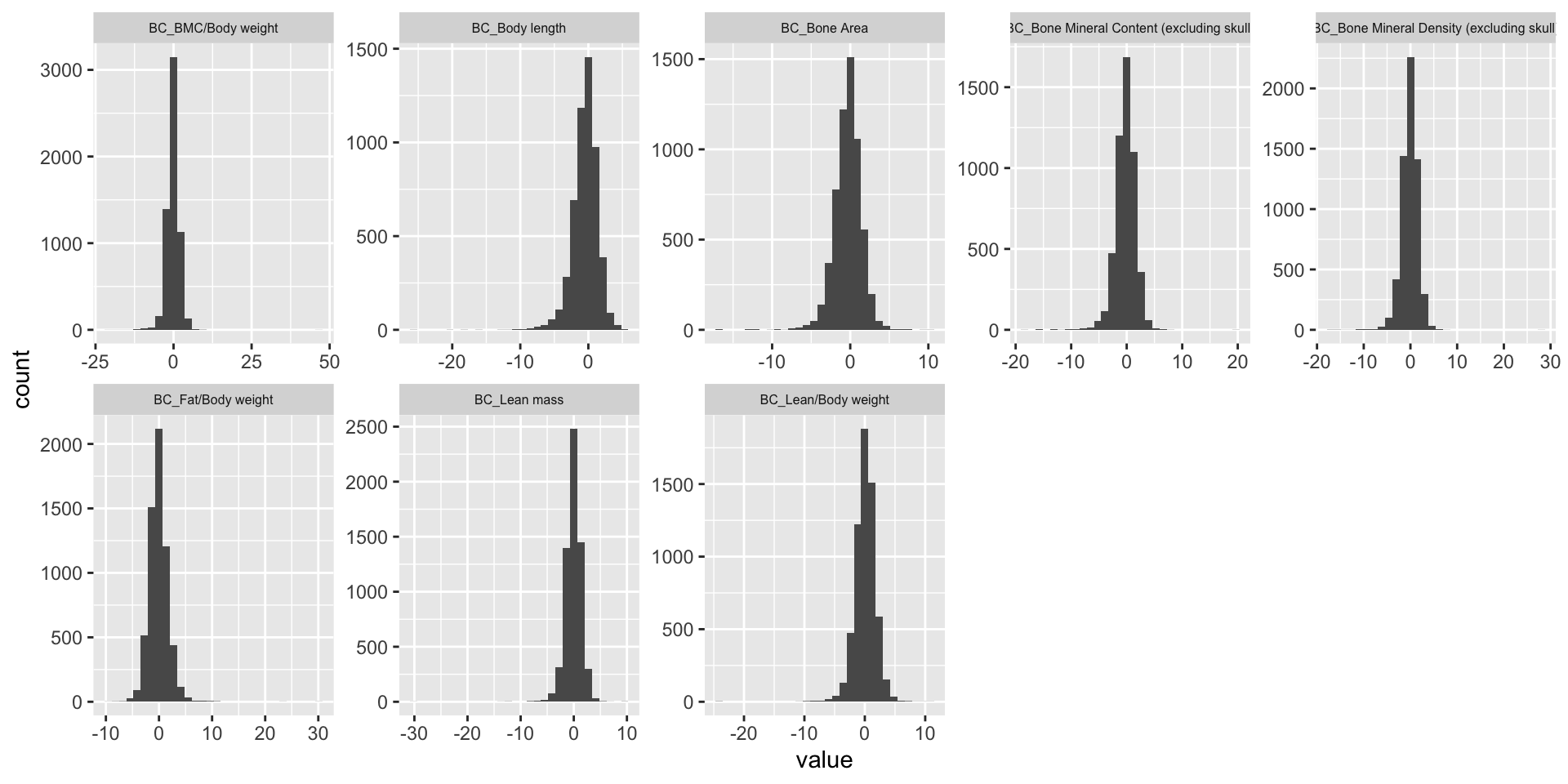
Estimation of Genetic Correlation Matrix Using Z-Scores
Here, we estimate the genetic correlations between phenotypes utilizing the association Z-score matrix.
# Select common phenotypes
BC.zmat <- BC.zmat[,common.pheno.list]
#dim(BC.zmat)
# Compute genetic correlations
BC.zcor = cor(BC.zmat, use="pairwise.complete.obs")
# Generate heatmap of the correlation matrix
ht = Heatmap(BC.zcor, cluster_rows = T, cluster_columns = T, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 10),
name="Genetic Corr (Z-score)"
)
draw(ht)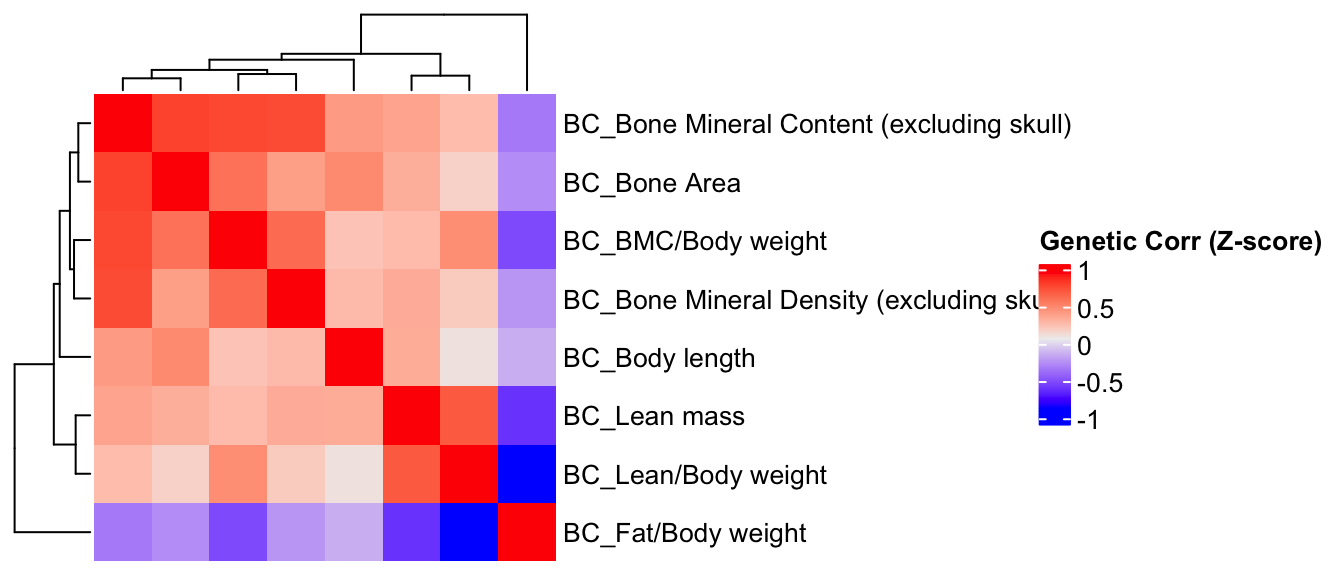
Comparison of Phenotypic Correlation and Genetic Correlation Among Phenotypes
We will compare the correlation matrix obtained from control mice phenotype data and the genetic correlation matrix estimated using association Z-scores. As depicted below, both correlation heatmaps show similar correlation patterns.
BC.cor.rank.fig <- BC.cor.rank[common.pheno.list,common.pheno.list]
BC.cor.fig <- BC.cor[common.pheno.list,common.pheno.list]
BC.cor.combat.fig <- BC.cor.combat[common.pheno.list, common.pheno.list]
BC.zcor.fig <- BC.zcor
ht = Heatmap(BC.cor.rank.fig, cluster_rows = TRUE, cluster_columns = TRUE, show_column_names = F, #col = col_fun,
show_row_dend = F, show_column_dend = F, # do not show dendrogram
row_names_gp = gpar(fontsize = 8), column_title="Phenotype Corr (RankZ, Pearson)", column_title_gp = gpar(fontsize = 8),
name="Corr")
pheno.order <- row_order(ht)
#draw(ht)
BC.cor.rank.fig <- BC.cor.rank.fig[pheno.order,pheno.order]
ht1 = Heatmap(BC.cor.rank.fig, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
show_row_dend = F, show_column_dend = F, # do not show dendrogram
row_names_gp = gpar(fontsize = 8), column_title="Phenotype Corr (RankZ, Pearson)", column_title_gp = gpar(fontsize = 8),
name="Corr")
BC.cor.fig <- BC.cor.fig[pheno.order,pheno.order]
ht2 = Heatmap(BC.cor.fig, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 8), column_title="Phenotype Corr (Spearman)", column_title_gp = gpar(fontsize = 8),
name="Corr")
BC.cor.combat.fig <- BC.cor.combat.fig[pheno.order,pheno.order]
ht3 = Heatmap(BC.cor.combat.fig, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 8), column_title="Phenotype Corr (Combat, Pearson)", column_title_gp = gpar(fontsize = 8),
name="Corr")
BC.zcor.fig <- BC.zcor.fig[pheno.order,pheno.order]
ht4 = Heatmap(BC.zcor.fig, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 8), column_title="Genetic Corr (Pearson)", column_title_gp = gpar(fontsize = 8),
name="Corr"
)
draw(ht1+ht2+ht3+ht4)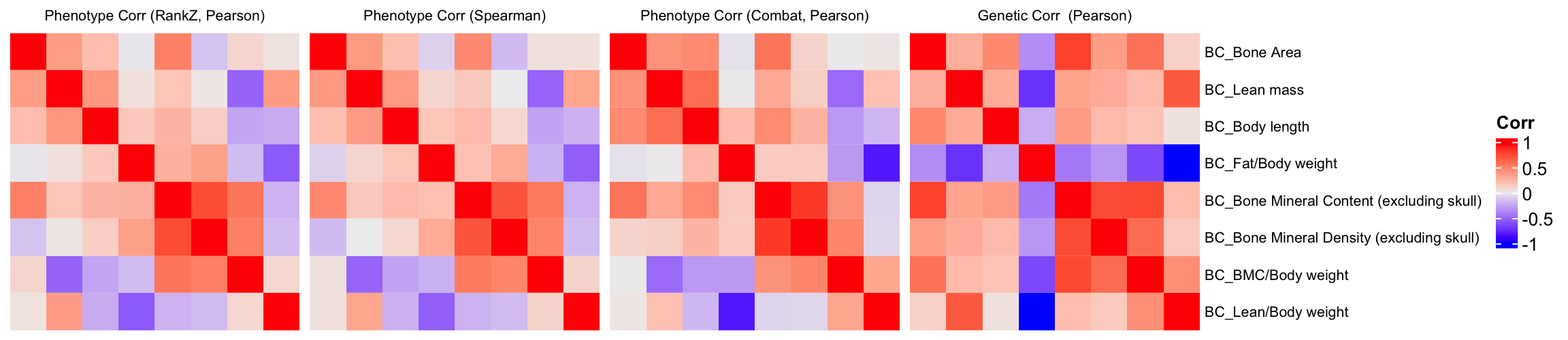
png(file="docs/figure/figures.Rmd/cors_BC_v16.png", width=800, height=250)
draw(ht1+ht2+ht3+ht4)
dev.off()quartz_off_screen
2 # Supplementary Figure 2
BC.cor.combat.fig2 <- BC.cor.combat.fig
BC.zcor.fig2 <- BC.zcor.fig
rownames(BC.cor.combat.fig2) <- substring(rownames(BC.cor.combat.fig2), 4)
rownames(BC.zcor.fig2) <- substring(rownames(BC.zcor.fig), 4)
rownames(BC.cor.combat.fig2)[c(6,7)] = c("Bone Mineral Content", "Bone Mineral Density")
rownames(BC.zcor.fig2)[c(6,7)] = c("Bone Mineral Content", "Bone Mineral Density")
ht3 = Heatmap(BC.cor.combat.fig2, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 12), column_title="Phenotypic Correlation", column_title_gp = gpar(fontsize = 15),
name="Corr",
heatmap_legend_param = list(title_gp=gpar(fontsize=15), labels_gp=gpar(fontsize=12),
legend_height=unit(50, "mm")))
ht4 = Heatmap(BC.zcor.fig2, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 12), column_title="Genetic Correlation", column_title_gp = gpar(fontsize = 15),
name="Corr")
png(file="docs/figure/figures.Rmd/cors_BC_supp_v16.png", width=625, height=250)
draw(ht4+ht3)
dev.off()quartz_off_screen
2 pdf(file="docs/figure/figures.Rmd/cors_BC_supp_v16.pdf", width=12, height=4.9)
draw(ht4+ht3)
dev.off()quartz_off_screen
2 Correlation Analysis Between Genetic Correlation Matrices Using Mantel’s Test
To evaluate the correlation between different genetic correlation matrices, we apply Mantel’s test, which measures the correlation between two distance matrices.
####################
# Use Mantel test
# https://stats.idre.ucla.edu/r/faq/how-can-i-perform-a-mantel-test-in-r/
# install.packages("ade4")
library(ade4)
# Function to extract upper triangular elements of a matrix
to.upper<-function(X) X[upper.tri(X,diag=FALSE)]
a1 <- to.upper(BC.cor.fig)
a2 <- to.upper(BC.cor.rank.fig)
a3 <- to.upper(BC.cor.combat.fig)
a4 <- to.upper(BC.zcor.fig)
plot(a4, a1)
| Version | Author | Date |
|---|---|---|
| 9b680ec | statsleelab | 2023-06-20 |
plot(a4, a2)
| Version | Author | Date |
|---|---|---|
| 9b680ec | statsleelab | 2023-06-20 |
plot(a4, a3)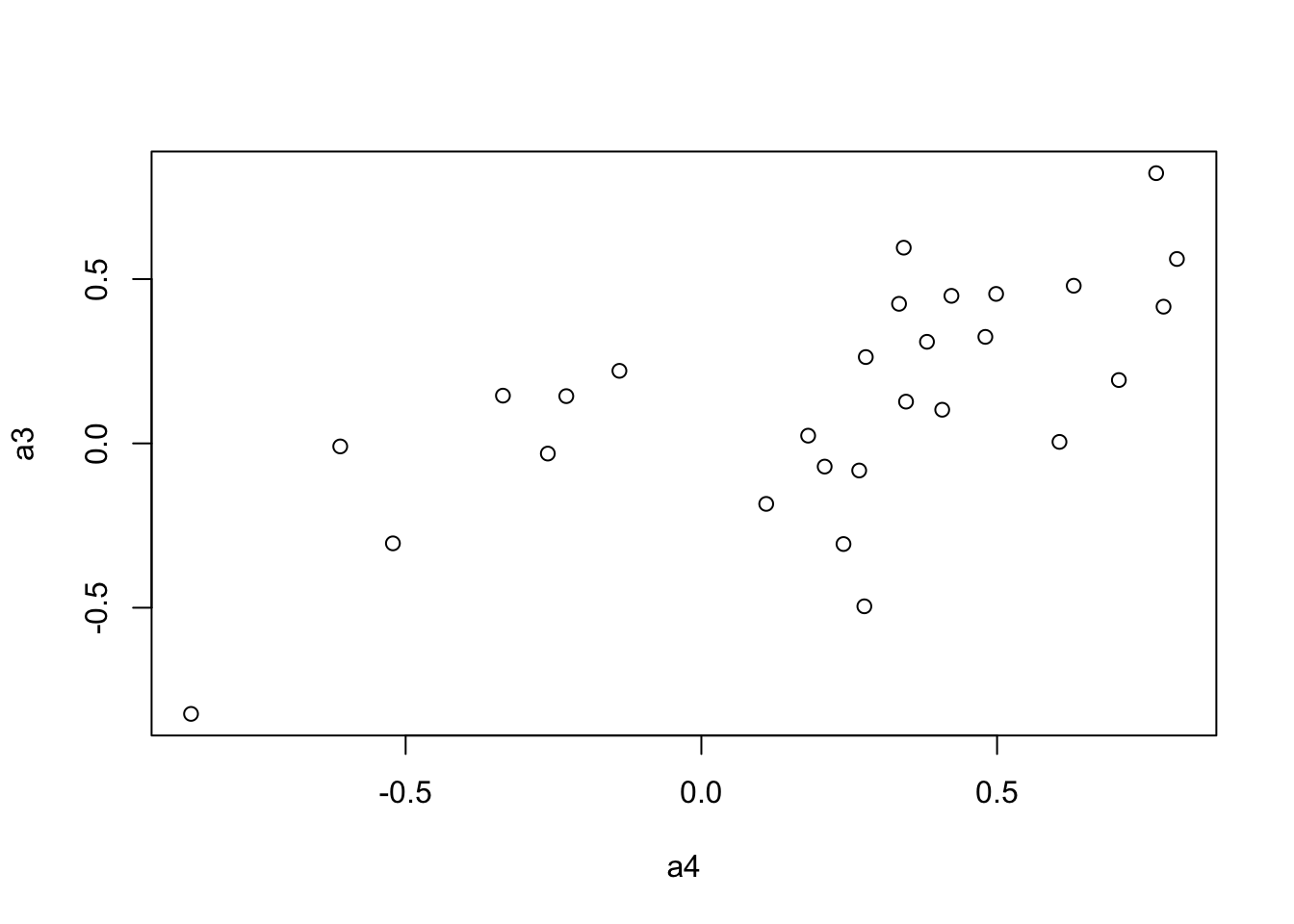
| Version | Author | Date |
|---|---|---|
| 9b680ec | statsleelab | 2023-06-20 |
mantel.rtest(as.dist(1-BC.cor.fig), as.dist(1-BC.zcor.fig), nrepet = 9999) #nrepet = number of permutationsMonte-Carlo test
Call: mantelnoneuclid(m1 = m1, m2 = m2, nrepet = nrepet)
Observation: 0.5396627
Based on 9999 replicates
Simulated p-value: 8e-04
Alternative hypothesis: greater
Std.Obs Expectation Variance
2.57683170 0.00431246 0.04316223 mantel.rtest(as.dist(1-BC.cor.rank.fig), as.dist(1-BC.zcor.fig), nrepet = 9999)Monte-Carlo test
Call: mantelnoneuclid(m1 = m1, m2 = m2, nrepet = nrepet)
Observation: 0.5383613
Based on 9999 replicates
Simulated p-value: 0.0013
Alternative hypothesis: greater
Std.Obs Expectation Variance
2.4666013697 0.0005465111 0.0475409136 mantel.rtest(as.dist(1-BC.cor.combat.fig), as.dist(1-BC.zcor.fig), nrepet = 9999)Monte-Carlo test
Call: mantelnoneuclid(m1 = m1, m2 = m2, nrepet = nrepet)
Observation: 0.6543789
Based on 9999 replicates
Simulated p-value: 2e-04
Alternative hypothesis: greater
Std.Obs Expectation Variance
2.608321834 -0.004513287 0.063812597 Evaluating the KOMPUTE Imputation Algorithm
Initializing the KOMPUTE Package
# Check if KOMPUTE is installed, if not, install it from GitHub using devtools
if(!"kompute" %in% rownames(installed.packages())){
library(devtools)
devtools::install_github("dleelab/kompute")
}
library(kompute)Simulation study - Comparison of imputed vs measured z-score values
In this section, we conduct a simulation study to compare the performance of the KOMPUTE method with the measured gene-phenotype association z-scores. We randomly select some of these measured z-scores, mask them, and then use the KOMPUTE method to impute them. We then compare the imputed z-scores with the measured ones.
zmat <-t(BC.zmat)
dim(zmat)[1] 8 6145# filter genes with less than 1 missing data point (na)
zmat0 <- is.na(zmat)
num.na<-colSums(zmat0)
#summary(num.na)
#dim(zmat)
#dim(zmat[,num.na<1])
#dim(zmat[,num.na<5])
#dim(zmat[,num.na<10])
# filter genes with less than 1 missing data point (na)
zmat <- zmat[,num.na<1]
#dim(zmat)
# Set correlation method for phenotypes
#pheno.cor <- BC.cor.fig
#pheno.cor <- BC.cor.rank.fig
pheno.cor <- BC.cor.combat.fig
#pheno.cor <- BC.zcor.fig
zmat <- zmat[rownames(pheno.cor),,drop=FALSE]
#rownames(zmat)
#rownames(pheno.cor)
#colnames(pheno.cor)
npheno <- nrow(zmat)
# calculate the percentage of missing Z-scores in the original data
100*sum(is.na(zmat))/(nrow(zmat)*ncol(zmat)) # 0%[1] 0nimp <- 1000 # # of missing/imputed Z-scores
set.seed(1234)
## find index of all measured zscores
all.i <- 1:(nrow(zmat)*ncol(zmat))
measured <- as.vector(!is.na(as.matrix(zmat)))
measured.i <- all.i[measured]
## mask 2000 measured z-scores
mask.i <- sort(sample(measured.i, nimp))
org.z = as.matrix(zmat)[mask.i]
zvec <- as.vector(as.matrix(zmat))
zvec[mask.i] <- NA
zmat.imp <- matrix(zvec, nrow=npheno)
rownames(zmat.imp) <- rownames(zmat)Run KOMPUTE method
kompute.res <- kompute(t(zmat.imp), pheno.cor, 0.01)
# Compare measured vs imputed z-scores
length(org.z)[1] 1000imp.z <- as.matrix(t(kompute.res$zmat))[mask.i]
imp.info <- as.matrix(t(kompute.res$infomat))[mask.i]
# Create a dataframe with the original and imputed z-scores and the information of imputed z-scores
imp <- data.frame(org.z=org.z, imp.z=imp.z, info=imp.info)
#dim(imp)
imp <- imp[complete.cases(imp),]
imp <- subset(imp, info>=0 & info <= 1)
#dim(imp)
cor.val <- round(cor(imp$imp.z, imp$org.z), digits=3)
#cor.val
g <- ggplot(imp, aes(x=imp.z, y=org.z)) +
geom_point() +
labs(title=paste0("IMPC BC Data, Cor=",cor.val),
x="Imputed Z-scores", y = "Measured Z-scores") +
theme_minimal()
g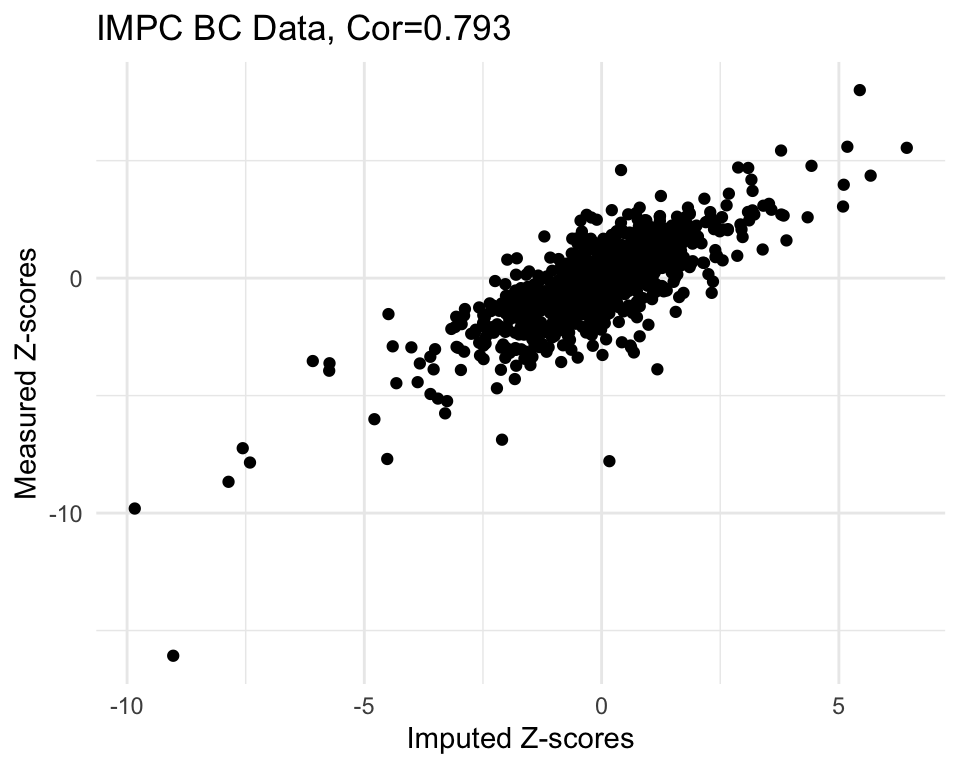
# Set a cutoff for information content and filter the data accordingly
info.cutoff <- 0.8
imp.sub <- subset(imp, info>info.cutoff)
#dim(imp.sub)
#summary(imp.sub$imp.z)
#summary(imp.sub$info)
cor.val <- round(cor(imp.sub$imp.z, imp.sub$org.z), digits=3)
#cor.val
g <- ggplot(imp.sub, aes(x=imp.z, y=org.z, col=info)) +
geom_point() +
labs(title=paste0("IMPC BC Data, Info>", info.cutoff, ", Cor=",cor.val),
x="Imputed Z-scores", y = "Measured Z-scores", col="Info") +
theme_minimal()
g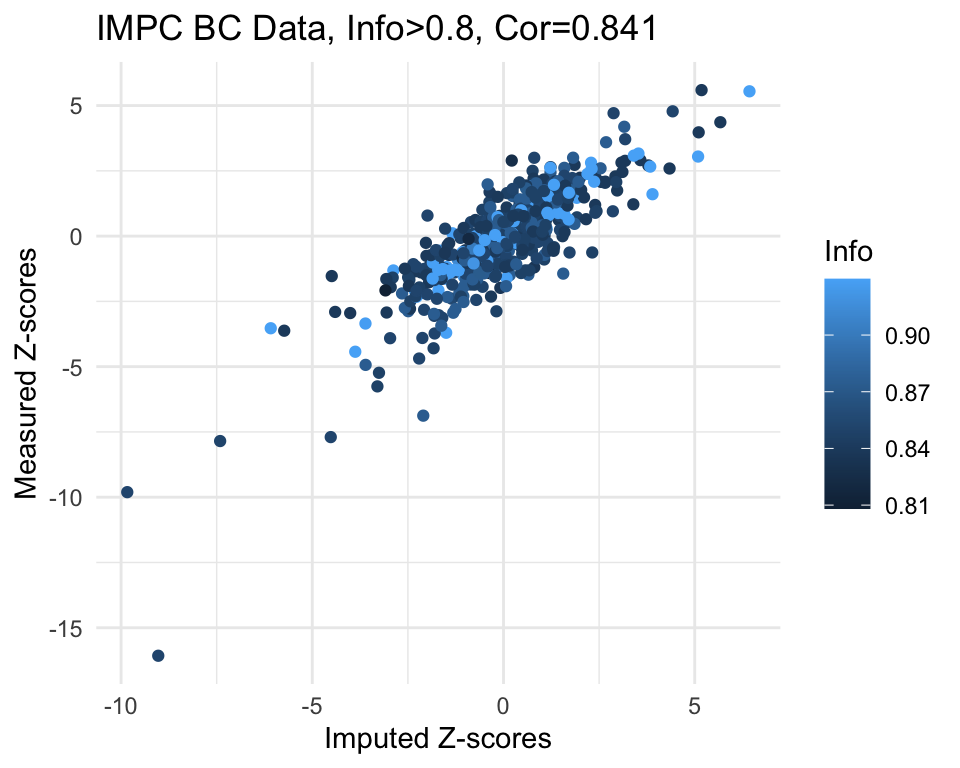
# save plot
png(file="docs/figure/figures.Rmd/sim_results_BC_v16.png", width=600, height=350)
g
dev.off()quartz_off_screen
2 # Part 1 of Figure 2
fig2.1 <- ggplot(imp.sub, aes(x=imp.z, y=org.z, col=info)) +
geom_point() +
labs(title="Body Composition",
x="Imputed Z-scores", y = "Measured Z-scores", col="Info") +
scale_x_continuous(limits=c(-9,9), breaks=c(seq(-9,9,3)), minor_breaks = NULL) +
scale_y_continuous(limits=c(-9,9), breaks=c(seq(-9,9,3))) +
scale_color_gradient(limits=c(0.8,1), low="#98cdf9", high="#084b82") +
theme_bw() +
theme(legend.position="none", plot.title=element_text(hjust=0.5))
save(fig2.1, file="docs/figure/figures.Rmd/sim_BC_v16.rdata")Run SVD Matrix Completion method
# load SVD Matrix Completion function
source("code/svd_impute.R")
r <- 6
mc.res <- svd.impute(zmat.imp, r)
# Compare measured vs imputed z-scores
length(org.z)[1] 1000imp.z <- mc.res[mask.i]
#plot(imp.z, org.z)
#cor(imp.z, org.z)
# Create a dataframe with the original and imputed z-scores and the information of imputed z-scores
imp2 <- data.frame(org.z=org.z, imp.z=imp.z)
#dim(imp2)
imp2 <- imp2[complete.cases(imp2),]
cor.val <- round(cor(imp2$imp.z, imp2$org.z), digits=3)
#cor.val
g <- ggplot(imp2, aes(x=imp.z, y=org.z)) +
geom_point() +
labs(title=paste0("IMPC BC Data, Cor=",cor.val),
x="Imputed Z-scores", y = "Measured Z-scores") +
theme_minimal()
g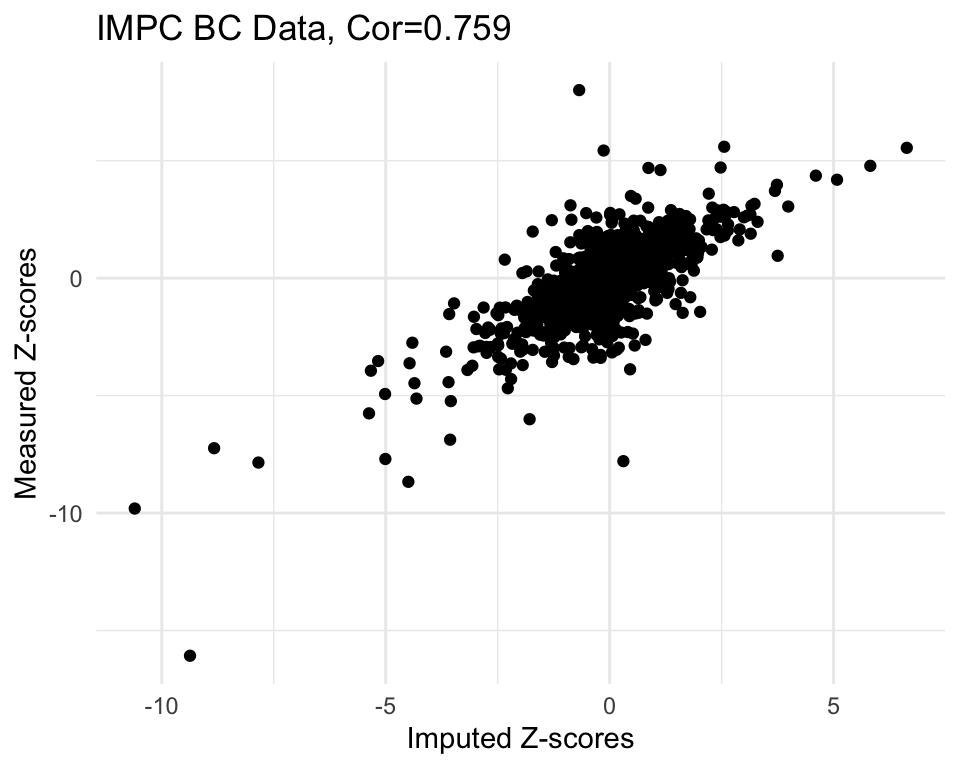
| Version | Author | Date |
|---|---|---|
| b343ff3 | statsleelab | 2023-07-03 |
Save imputation results
imp$method <- "KOMPUTE"
imp2$method <- "SVD-MC"
imp2$info <- NA
BC_Imputation_Result <- rbind(imp, imp2)
save(BC_Imputation_Result, file = "data/BC.imp.res.v16.RData")Save z-score matrix and phenotype correlation matrix
plist <- sort(colnames(BC.zmat))
BC_Zscore_Mat <- as.matrix(BC.zmat[,plist])
save(BC_Zscore_Mat, file = "data/BC.zmat.v16.RData")
BC_Pheno_Cor <- pheno.cor[plist,plist]
save(BC_Pheno_Cor, file = "data/BC.pheno.cor.v16.RData")
sessionInfo()R version 4.2.1 (2022-06-23)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.7
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] kompute_0.1.0 ade4_1.7-20 sva_3.44.0
[4] BiocParallel_1.30.3 genefilter_1.78.0 mgcv_1.8-40
[7] nlme_3.1-158 lme4_1.1-31 Matrix_1.5-1
[10] RNOmni_1.0.1 ggpubr_0.5.0 patchwork_1.1.2
[13] ComplexHeatmap_2.12.1 circlize_0.4.15 RColorBrewer_1.1-3
[16] tidyr_1.2.0 ggplot2_3.4.1 reshape2_1.4.4
[19] dplyr_1.0.9 data.table_1.14.2 workflowr_1.7.0.1
loaded via a namespace (and not attached):
[1] minqa_1.2.5 colorspace_2.1-0 ggsignif_0.6.4
[4] rjson_0.2.21 rprojroot_2.0.3 XVector_0.36.0
[7] GlobalOptions_0.1.2 fs_1.5.2 clue_0.3-62
[10] rstudioapi_0.13 farver_2.1.1 bit64_4.0.5
[13] AnnotationDbi_1.58.0 fansi_1.0.4 codetools_0.2-18
[16] splines_4.2.1 doParallel_1.0.17 cachem_1.0.6
[19] knitr_1.39 jsonlite_1.8.0 nloptr_2.0.3
[22] broom_1.0.1 annotate_1.74.0 cluster_2.1.3
[25] png_0.1-8 compiler_4.2.1 httr_1.4.3
[28] backports_1.4.1 assertthat_0.2.1 fastmap_1.1.0
[31] limma_3.52.4 cli_3.6.0 later_1.3.0
[34] htmltools_0.5.3 tools_4.2.1 GenomeInfoDbData_1.2.8
[37] gtable_0.3.1 glue_1.6.2 Rcpp_1.0.10
[40] carData_3.0-5 Biobase_2.56.0 jquerylib_0.1.4
[43] vctrs_0.5.2 Biostrings_2.64.0 iterators_1.0.14
[46] xfun_0.31 stringr_1.4.0 ps_1.7.1
[49] lifecycle_1.0.3 rstatix_0.7.1 XML_3.99-0.10
[52] edgeR_3.38.4 zlibbioc_1.42.0 getPass_0.2-2
[55] MASS_7.3-58.1 scales_1.2.1 promises_1.2.0.1
[58] parallel_4.2.1 yaml_2.3.5 memoise_2.0.1
[61] sass_0.4.2 stringi_1.7.8 RSQLite_2.2.15
[64] highr_0.9 S4Vectors_0.34.0 foreach_1.5.2
[67] BiocGenerics_0.42.0 boot_1.3-28 shape_1.4.6
[70] GenomeInfoDb_1.32.3 rlang_1.0.6 pkgconfig_2.0.3
[73] matrixStats_0.62.0 bitops_1.0-7 evaluate_0.16
[76] lattice_0.20-45 purrr_0.3.4 labeling_0.4.2
[79] cowplot_1.1.1 bit_4.0.4 processx_3.7.0
[82] tidyselect_1.2.0 plyr_1.8.7 magrittr_2.0.3
[85] R6_2.5.1 IRanges_2.30.0 generics_0.1.3
[88] DBI_1.1.3 pillar_1.8.1 whisker_0.4
[91] withr_2.5.0 survival_3.3-1 KEGGREST_1.36.3
[94] abind_1.4-5 RCurl_1.98-1.8 tibble_3.1.8
[97] crayon_1.5.1 car_3.1-1 utf8_1.2.3
[100] rmarkdown_2.14 GetoptLong_1.0.5 locfit_1.5-9.6
[103] blob_1.2.3 callr_3.7.1 git2r_0.30.1
[106] digest_0.6.29 xtable_1.8-4 httpuv_1.6.5
[109] stats4_4.2.1 munsell_0.5.0 bslib_0.4.0