KOMPUTE method testing - CC data (v10.1)
Coby Warkentin and Donghyung Lee
2023-06-19
- Load packages
- Prep control phenotype data
- Load Clinical Chemistry control phenotypes
- Heatmap showing measured phenotypes
- Remove phenotypes with num of obs < 20000
- Romove samples with num of measured phenotypes < 15
- Heapmap of measured phenotypes after filtering
- Reshape the data (long to wide)
- Distribution of each phenotype
- Rank Z transformation
- Principal Variance Component Analysis
- Removing batch effects using ComBat
- PVCA on residuals from ComBat
- Compute correlations between phenotypes
- Prep IMPC summary stat
- Read CC summary stat (IMPCv10.1)
- Duplicates in gene-phenotype pair
- Using Stouffer’s method, merge multiple z-scores of a gene-phenotype pair into a z-score
- Make z-score matrix (long to wide)
- Z-score Distribution
- Estimate genetic correlation matrix between phenotypes using Zscores
- Phenotype Corr VS Genetic Corr btw phenotypes
- Test of the correlation between genetic correlation matrices
- Test KOMPUTE imputation algorithm
Last updated: 2023-06-19
Checks: 7 0
Knit directory: komputeExamples/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230110) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version bb7f5ee. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: code/.DS_Store
Untracked files:
Untracked: analysis/figures.Rmd_old
Unstaged changes:
Deleted: analysis/figures.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/kompute_test_CC_v10.1.Rmd)
and HTML (docs/kompute_test_CC_v10.1.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 7685a09 | statsleelab | 2023-01-10 | first commit |
| html | 7685a09 | statsleelab | 2023-01-10 | first commit |
Load packages
rm(list=ls())
library(data.table)
library(dplyr)
library(reshape2)
library(ggplot2)
library(tidyr) #spread
library(RColorBrewer)
#library(irlba) # partial PCA
#library(cowplot)
library(circlize)
library(ComplexHeatmap)Prep control phenotype data
Load Clinical Chemistry control phenotypes
CC.data <- readRDS("data/CC.data.rds")
dim(CC.data)[1] 679047 10Heatmap showing measured phenotypes
This heatmaps show phenotypes measured for each control mouse. Columns represent mice and rows represent phenotypes.
mtest <- table(CC.data$proc_param_name_stable_id, CC.data$biological_sample_id)
mtest <-as.data.frame.matrix(mtest)
dim(mtest)[1] 155 34952if(FALSE){
nmax <-max(mtest)
library(circlize)
col_fun = colorRamp2(c(0, nmax), c("white", "red"))
col_fun(seq(0, nmax))
ht = Heatmap(as.matrix(mtest), cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, col = col_fun,
row_names_gp = gpar(fontsize = 8), name="Count")
draw(ht)
}Remove phenotypes with num of obs < 20000
mtest <- table(CC.data$proc_param_name, CC.data$biological_sample_id)
dim(mtest)[1] 36 34952#head(mtest[,1:10])
mtest0 <- mtest>0
#head(mtest0[,1:10])
rowSums(mtest0) CC_Alanine aminotransferase
34633
CC_Albumin
34594
CC_Alkaline phosphatase
34450
CC_Alpha-amylase
22077
CC_Aspartate aminotransferase
34078
CC_Calcium
34526
CC_Chloride
24604
CC_Cholesterol ratio
25
CC_Creatine kinase
15761
CC_Creatinine
29741
CC_Ferretin
150
CC_Free fatty acid
4338
CC_Free fatty acids
6147
CC_Fructosamine
12867
CC_Glucose
34118
CC_Glycerol
7509
CC_Glycosilated hemoglobin A1c (HbA1c)
1698
CC_HDL-cholesterol
28478
CC_Iron
25602
CC_Lactate dehydrogenase
8967
CC_LDL-cholesterol
11387
CC_Lipase
2777
CC_Magnesium
7682
CC_Phosphorus
34361
CC_Potassium
24401
CC_Sodium
24609
CC_Thyroxine
3540
CC_Total bilirubin
29694
CC_Total cholesterol
34394
CC_Total protein
34429
CC_Transferrin
169
CC_Triglycerides
33795
CC_UIBC (unsaturated iron binding capacity)
2801
CC_Urea
8296
CC_Urea (Blood Urea Nitrogen - BUN)
26272
CC_Uric acid
4299 rmv.pheno.list <- rownames(mtest)[rowSums(mtest0)<20000]
#rmv.pheno.list
dim(CC.data)[1] 679047 10CC.data <- CC.data %>% filter(!(proc_param_name %in% rmv.pheno.list))
dim(CC.data)[1] 580566 10# number of phenotypes left
length(unique(CC.data$proc_param_name))[1] 19Romove samples with num of measured phenotypes < 15
mtest <- table(CC.data$proc_param_name, CC.data$biological_sample_id)
dim(mtest)[1] 19 34925head(mtest[,1:10])
21 22 24 25 26 27 28 29 30 31
CC_Alanine aminotransferase 1 1 1 1 1 1 1 0 1 1
CC_Albumin 1 1 1 1 1 1 1 0 1 1
CC_Alkaline phosphatase 1 1 1 1 1 1 1 0 1 1
CC_Alpha-amylase 1 1 1 1 1 1 1 0 1 1
CC_Aspartate aminotransferase 1 1 1 1 1 1 1 1 1 1
CC_Calcium 1 1 1 1 1 1 1 1 1 1mtest0 <- mtest>0
head(mtest0[,1:10])
21 22 24 25 26 27 28 29 30
CC_Alanine aminotransferase TRUE TRUE TRUE TRUE TRUE TRUE TRUE FALSE TRUE
CC_Albumin TRUE TRUE TRUE TRUE TRUE TRUE TRUE FALSE TRUE
CC_Alkaline phosphatase TRUE TRUE TRUE TRUE TRUE TRUE TRUE FALSE TRUE
CC_Alpha-amylase TRUE TRUE TRUE TRUE TRUE TRUE TRUE FALSE TRUE
CC_Aspartate aminotransferase TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
CC_Calcium TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
31
CC_Alanine aminotransferase TRUE
CC_Albumin TRUE
CC_Alkaline phosphatase TRUE
CC_Alpha-amylase TRUE
CC_Aspartate aminotransferase TRUE
CC_Calcium TRUEsummary(colSums(mtest0)) Min. 1st Qu. Median Mean 3rd Qu. Max.
1.00 15.00 17.00 16.57 19.00 19.00 rmv.sample.list <- colnames(mtest)[colSums(mtest0)<15]
length(rmv.sample.list)[1] 8380dim(CC.data)[1] 580566 10CC.data <- CC.data %>% filter(!(biological_sample_id %in% rmv.sample.list))
dim(CC.data)[1] 472110 10# number of observations to use
length(unique(CC.data$biological_sample_id))[1] 26545Heapmap of measured phenotypes after filtering
if(FALSE){
mtest <- table(CC.data$proc_param_name, CC.data$biological_sample_id)
dim(mtest)
mtest <-as.data.frame.matrix(mtest)
nmax <-max(mtest)
library(circlize)
col_fun = colorRamp2(c(0, nmax), c("white", "red"))
col_fun(seq(0, nmax))
pdf("~/Google Drive Miami/Miami_IMPC/output/measured_phenotypes_controls_after_filtering_CC.pdf", width = 10, height = 3)
ht = Heatmap(as.matrix(mtest), cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, col = col_fun,
row_names_gp = gpar(fontsize = 7), name="Count")
draw(ht)
dev.off()
}Reshape the data (long to wide)
CC.mat <- CC.data %>%
dplyr::select(biological_sample_id, proc_param_name, data_point, sex, phenotyping_center, strain_name) %>%
##consider weight or age in weeks
arrange(biological_sample_id) %>%
distinct(biological_sample_id, proc_param_name, .keep_all=TRUE) %>% ## remove duplicates, maybe mean() is better.
spread(proc_param_name, data_point) %>%
tibble::column_to_rownames(var="biological_sample_id")
head(CC.mat) sex phenotyping_center strain_name CC_Alanine aminotransferase
21 male MRC Harwell 129S8/SvEv-Gpi1<c>/NimrH 85.9
22 male MRC Harwell 129S8/SvEv-Gpi1<c>/NimrH 110.9
24 male MRC Harwell 129S8/SvEv-Gpi1<c>/NimrH 32.1
25 male MRC Harwell 129S8/SvEv-Gpi1<c>/NimrH 33.7
26 male MRC Harwell 129S8/SvEv-Gpi1<c>/NimrH 37.2
27 male MRC Harwell 129S8/SvEv-Gpi1<c>/NimrH 39.7
CC_Albumin CC_Alkaline phosphatase CC_Alpha-amylase
21 25.3 90 759.2
22 26.9 86 844.3
24 26.5 103 822.9
25 26.2 81 799.9
26 28.4 95 810.5
27 27.3 93 821.4
CC_Aspartate aminotransferase CC_Calcium CC_Chloride CC_Creatinine
21 97.7 2.33 112 NA
22 114.7 2.41 113 NA
24 57.7 2.35 108 NA
25 64.0 2.35 110 NA
26 62.3 2.35 109 NA
27 58.3 2.37 109 NA
CC_Glucose CC_HDL-cholesterol CC_Iron CC_Phosphorus CC_Potassium CC_Sodium
21 8.46 NA 37.86 1.76 4.8 152
22 9.83 NA 39.78 1.82 5.7 153
24 8.36 NA 38.24 1.89 4.7 154
25 10.42 NA 36.28 2.10 4.8 153
26 9.79 NA 36.26 2.02 5.1 153
27 9.74 NA 38.30 1.57 4.5 153
CC_Total bilirubin CC_Total cholesterol CC_Total protein CC_Triglycerides
21 NA 3.27 50.6 1.04
22 NA 3.40 52.4 1.02
24 NA 3.63 52.4 1.43
25 NA 3.40 51.6 0.72
26 NA 3.53 51.9 1.15
27 NA 3.20 51.8 1.12
CC_Urea (Blood Urea Nitrogen - BUN)
21 NA
22 NA
24 NA
25 NA
26 NA
27 NAdim(CC.mat)[1] 26545 22summary(colSums(is.na(CC.mat[,-1:-3]))) Min. 1st Qu. Median Mean 3rd Qu. Max.
25 61 134 1784 2930 7853 Distribution of each phenotype
ggplot(melt(CC.mat), aes(x=value)) +
geom_histogram() +
facet_wrap(~variable, scales="free", ncol=5)+
theme(strip.text.x = element_text(size = 6))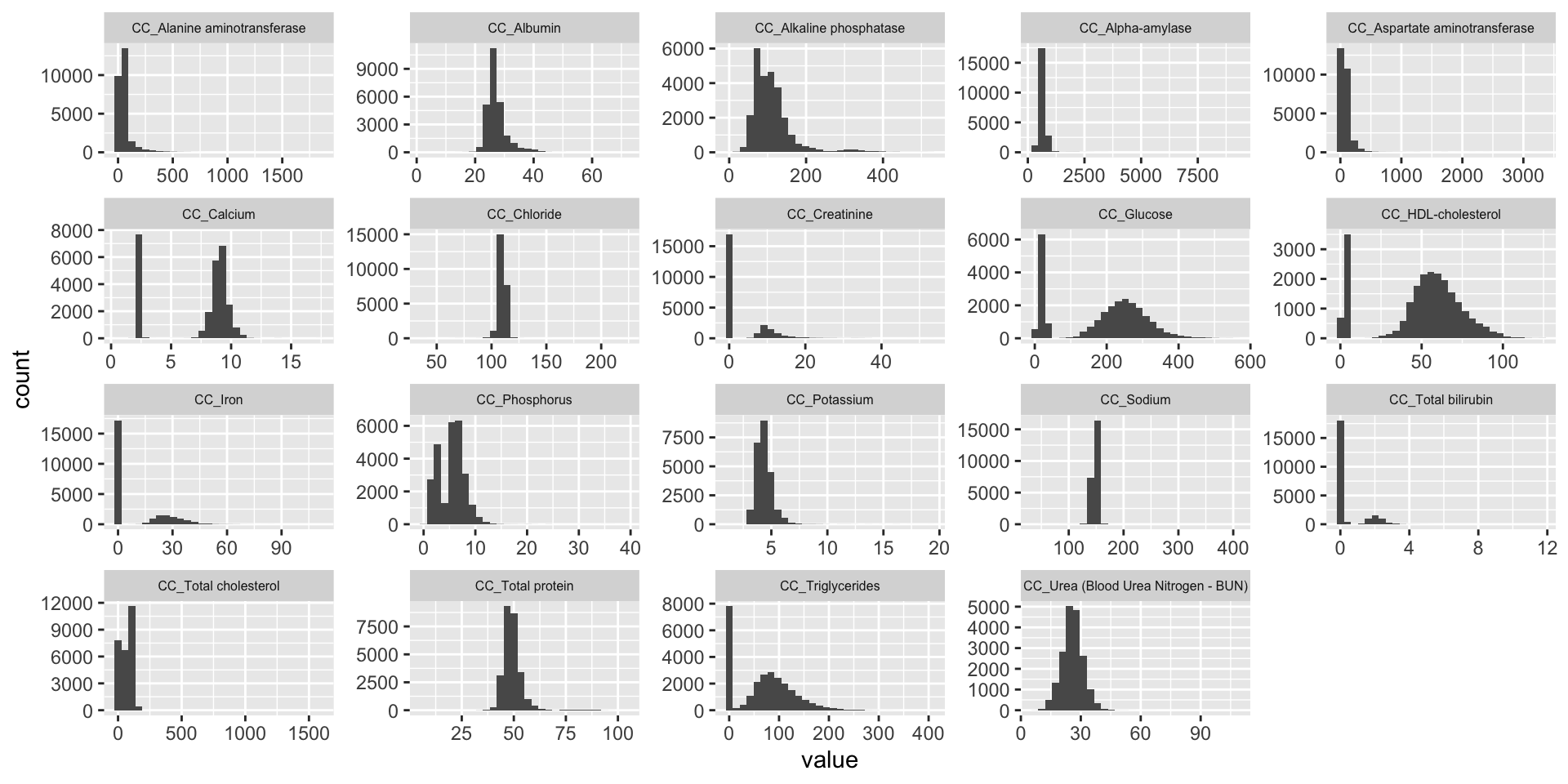
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Rank Z transformation
library(RNOmni)
CC.mat.rank <- CC.mat
dim(CC.mat.rank)[1] 26545 22CC.mat.rank <- CC.mat.rank[complete.cases(CC.mat.rank),]
dim(CC.mat.rank)[1] 11663 22dim(CC.mat)[1] 26545 22CC.mat <- CC.mat[complete.cases(CC.mat),]
dim(CC.mat)[1] 11663 22CC.mat.rank <- cbind(CC.mat.rank[,1:3], apply(CC.mat.rank[,-1:-3], 2, RankNorm))
ggplot(melt(CC.mat.rank), aes(x=value)) +
geom_histogram() +
facet_wrap(~variable, scales="free", ncol=5)+
theme(strip.text.x = element_text(size = 6))![]()
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Principal Variance Component Analysis
Here we conducted a PVCA analysis on the phenotype matrix data and measure the proportion of variance explained by each important covariate (sex, phenotyping_center).
source("code/PVCA.R")
meta <- CC.mat.rank[,1:3] ## looking at covariates sex, phenotyping_center, and strain_name
head(meta) sex phenotyping_center strain_name
39638 female MRC Harwell C57BL/6NTac
39639 female HMGU C57BL/6NCrl
39640 female HMGU C57BL/6NTac
39643 female HMGU C57BL/6NCrl
39650 female HMGU C57BL/6NTac
39657 male HMGU C57BL/6NCrldim(meta)[1] 11663 3summary(meta) # variables are still characters sex phenotyping_center strain_name
Length:11663 Length:11663 Length:11663
Class :character Class :character Class :character
Mode :character Mode :character Mode :character meta[sapply(meta, is.character)] <- lapply(meta[sapply(meta, is.character)], as.factor)
summary(meta) # now all variables are converted to factors sex phenotyping_center strain_name
female:5850 HMGU :2552 B6Brd;B6Dnk;B6N-Tyr<c-Brd>: 164
male :5813 MRC Harwell:4801 C57BL/6N :4146
WTSI :4310 C57BL/6NCrl : 891
C57BL/6NTac :6462 chisq.test(meta[,1],meta[,2])
Pearson's Chi-squared test
data: meta[, 1] and meta[, 2]
X-squared = 0.032984, df = 2, p-value = 0.9836chisq.test(meta[,2],meta[,3])
Pearson's Chi-squared test
data: meta[, 2] and meta[, 3]
X-squared = 14688, df = 6, p-value < 2.2e-16meta<-meta[,-3] # phenotyping_center and strain_name strongly associated and this caused confouding in PVCA analysis so strain_name dropped.
G <- t(CC.mat.rank[,-1:-3]) ## phenotype matrix data
set.seed(09302021)
# Perform PVCA for 10 random samples of size 1000 (more computationally efficient)
pvca.res <- matrix(nrow=10, ncol=3)
for (i in 1:10){
sample <- sample(1:ncol(G), 1000, replace=FALSE)
pvca.res[i,] <- PVCA(G[,sample], meta[sample,], threshold=0.6, inter=FALSE)
}
# Average effect size across samples
pvca.means <- colMeans(pvca.res)
names(pvca.means) <- c(colnames(meta), "resid")
# Plot PVCA
pvca.plot <- PlotPVCA(pvca.means, "PVCA of Phenotype Matrix Data")
pvca.plot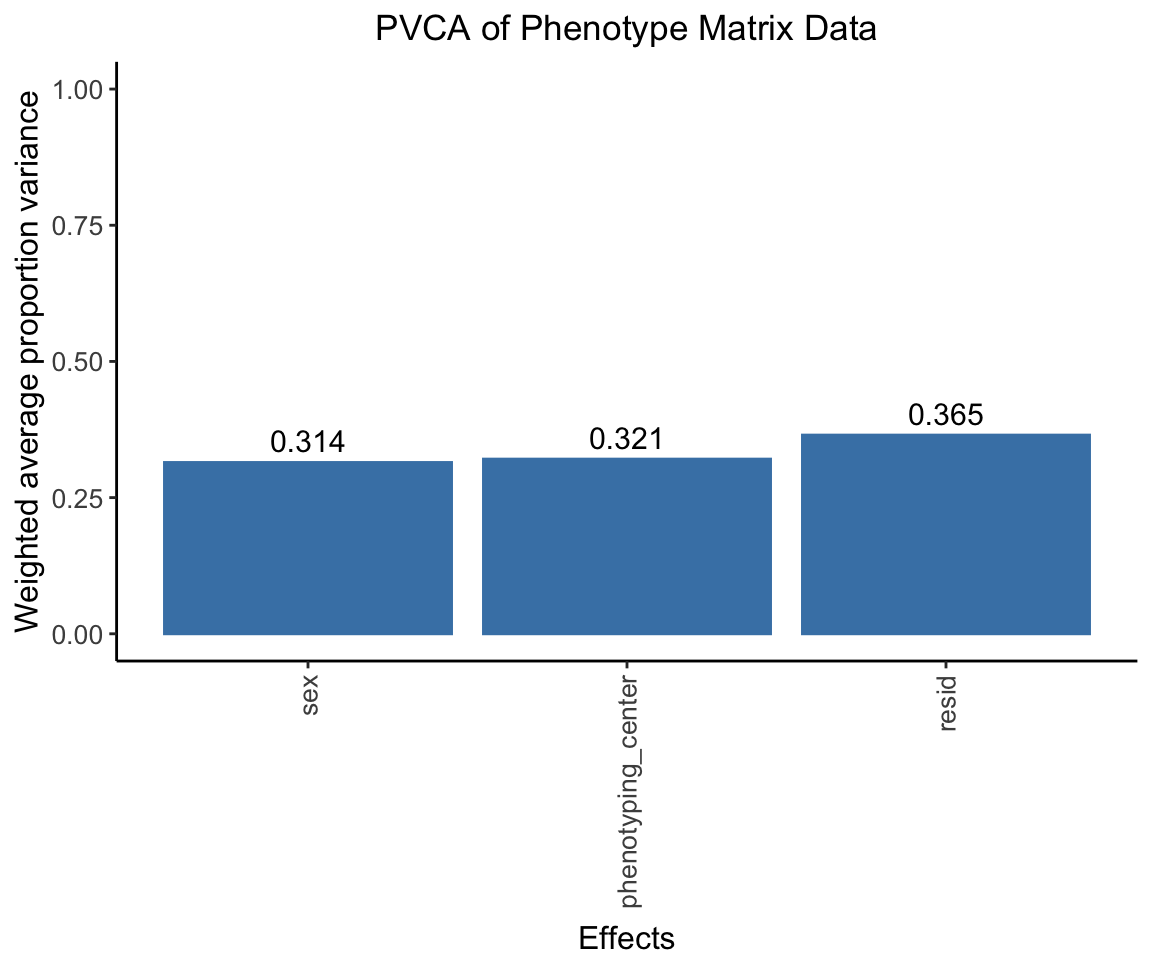
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
png(file="docs/figure/figures.Rmd/pvca_CC_1.png", width=600, height=350)
pvca.plot
dev.off()quartz_off_screen
2 Removing batch effects using ComBat
We remove the center effect using ComBat.
library(sva)Loading required package: mgcvLoading required package: nlme
Attaching package: 'nlme'The following object is masked from 'package:lme4':
lmListThe following object is masked from 'package:dplyr':
collapseThis is mgcv 1.8-40. For overview type 'help("mgcv-package")'.Loading required package: genefilter
Attaching package: 'genefilter'The following object is masked from 'package:ComplexHeatmap':
dist2Loading required package: BiocParallelcombat_komp = ComBat(dat=G, batch=meta$phenotyping_center, par.prior=TRUE, prior.plots=TRUE, mod=NULL)Found3batchesAdjusting for0covariate(s) or covariate level(s)Standardizing Data across genesFitting L/S model and finding priorsFinding parametric adjustmentsAdjusting the Data
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
combat_komp[1:5,1:5] 39638 39639 39640 39643
CC_Alanine aminotransferase 0.9340988 -0.5544734 0.6666494 -0.9010928
CC_Albumin 2.5845338 1.2282189 0.2400336 0.5834969
CC_Alkaline phosphatase 1.8038375 0.7077080 0.6239891 0.4510424
CC_Alpha-amylase 0.3151157 -0.3139508 0.3021379 0.3236144
CC_Aspartate aminotransferase 0.9536882 0.2034362 1.0860320 0.5575948
39650
CC_Alanine aminotransferase -1.1876876
CC_Albumin -0.4044527
CC_Alkaline phosphatase 0.2895801
CC_Alpha-amylase -1.4941108
CC_Aspartate aminotransferase -2.4362255G[1:5,1:5] # for comparison, combat_komp is same form and same dimensions as G 39638 39639 39640 39643
CC_Alanine aminotransferase 0.6215108 -1.3455580 0.2437437 -1.7966860
CC_Albumin 2.8129728 2.0400251 0.9990476 1.3608599
CC_Alkaline phosphatase 1.8346659 1.1364171 1.0485945 0.8671703
CC_Alpha-amylase 0.3053479 -0.4417481 0.1869870 0.2089043
CC_Aspartate aminotransferase 0.5629829 0.1714807 1.1471404 0.5629829
39650
CC_Alanine aminotransferase -2.1696915
CC_Albumin 0.3201306
CC_Alkaline phosphatase 0.6977934
CC_Alpha-amylase -1.6461331
CC_Aspartate aminotransferase -2.7465161PVCA on residuals from ComBat
The center effect should be much lower.
set.seed(09302021)
# Perform PVCA for 10 samples (more computationally efficient)
pvca.res.nobatch <- matrix(nrow=10, ncol=3)
for (i in 1:10){
sample <- sample(1:ncol(combat_komp), 1000, replace=FALSE)
pvca.res.nobatch[i,] <- PVCA(combat_komp[,sample], meta[sample,], threshold=0.6, inter=FALSE)
}
# Average effect size across samples
pvca.means.nobatch <- colMeans(pvca.res.nobatch)
names(pvca.means.nobatch) <- c(colnames(meta), "resid")
# Plot PVCA
pvca.plot.nobatch <- PlotPVCA(pvca.means.nobatch, "PVCA of Phenotype Matrix Data with Reduced Batch Effect")
pvca.plot.nobatch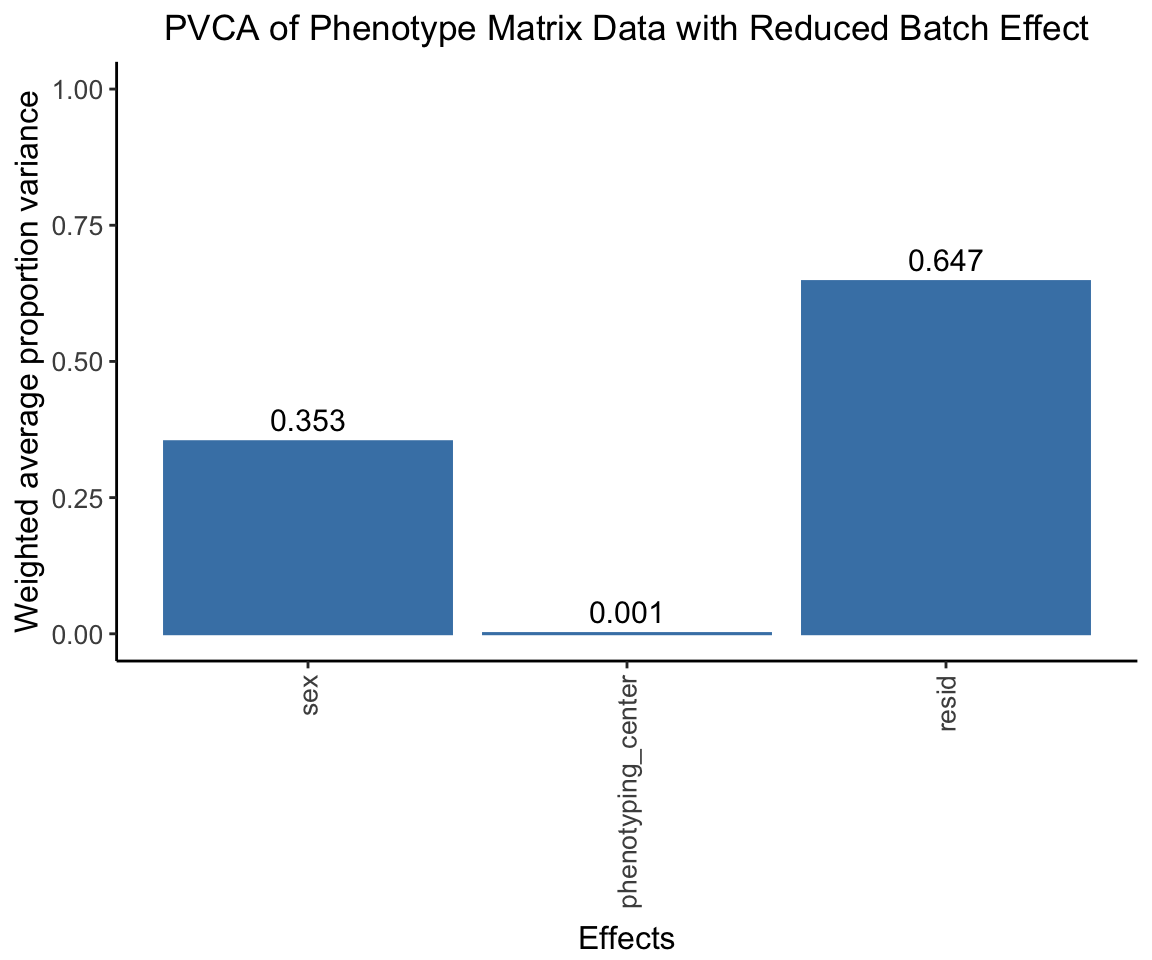
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
png(file="docs/figure/figures.Rmd/pvca_CC_2.png", width=600, height=350)
pvca.plot.nobatch
dev.off()quartz_off_screen
2 Compute correlations between phenotypes
CC.cor.rank <- cor(CC.mat.rank[,-1:-3], use="pairwise.complete.obs") # pearson correlation coefficient
CC.cor <- cor(CC.mat[,-1:-3], use="pairwise.complete.obs", method="spearman") # spearman
CC.cor.combat <- cor(t(combat_komp), use="pairwise.complete.obs")
pheno.list <- rownames(CC.cor)
ht1 = Heatmap(CC.cor, show_column_names = F, row_names_gp = gpar(fontsize = 9), name="Spearm. Corr.")
draw(ht1)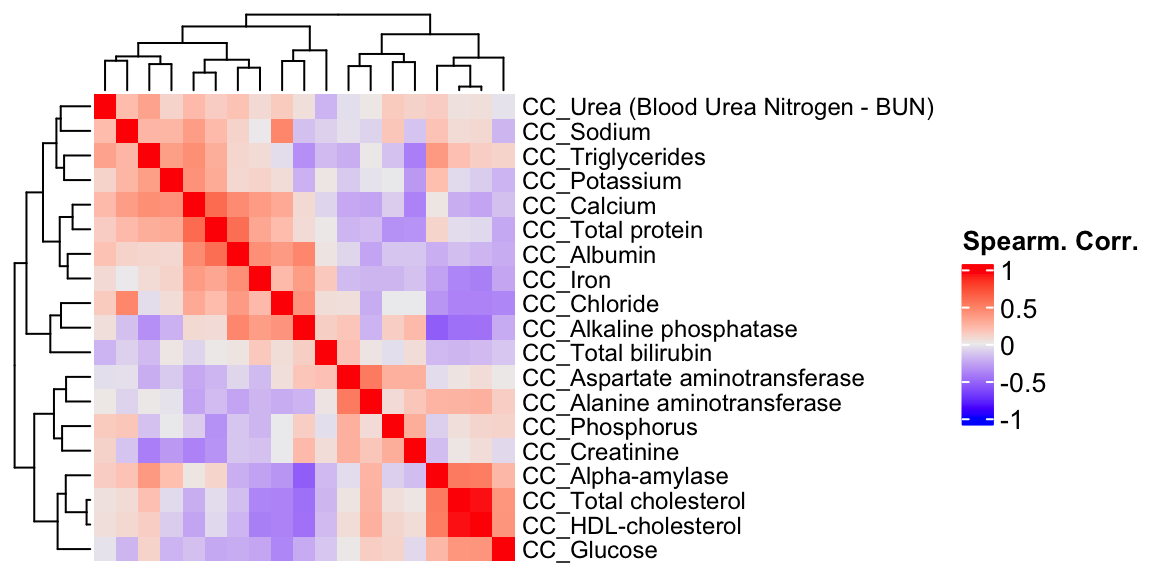
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
ht2 = Heatmap(CC.cor.rank, show_column_names = F, row_names_gp = gpar(fontsize = 9), name="Corr. RankZ")
draw(ht2)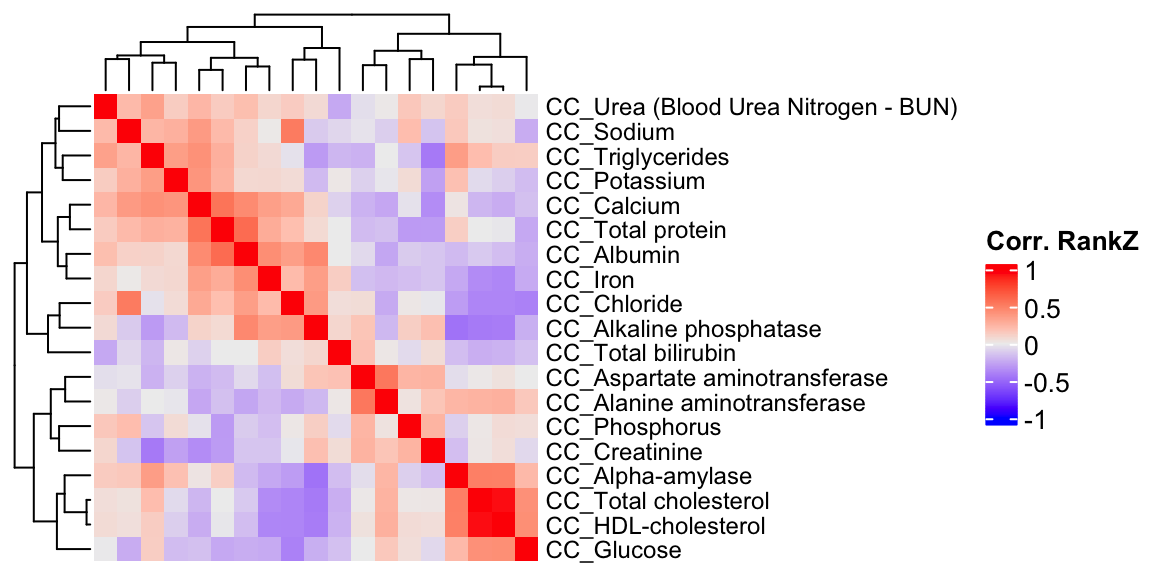
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
ht3 = Heatmap(CC.cor.combat, show_column_names = F, row_names_gp = gpar(fontsize = 9), name="Corr. ComBat")
draw(ht3)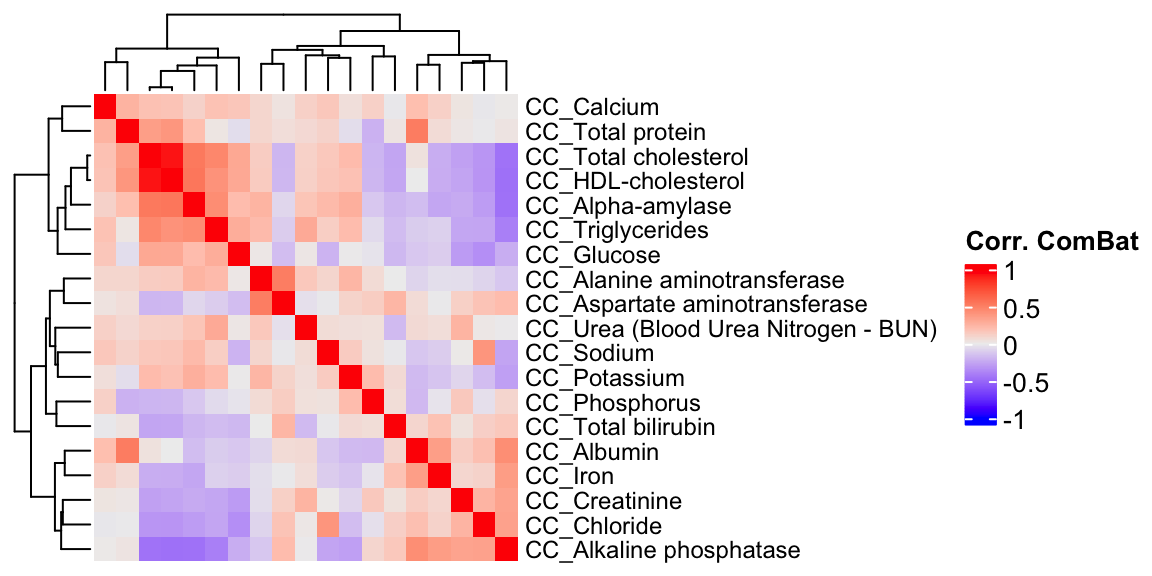
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Prep IMPC summary stat
Read CC summary stat (IMPCv10.1)
CC.stat <- readRDS("data/CC.stat.rds")
dim(CC.stat)[1] 95963 8table(CC.stat$parameter_name, CC.stat$procedure_name)
CC
Alanine aminotransferase 5005
Albumin 5007
Alkaline phosphatase 4987
Alpha-amylase 3112
Aspartate aminotransferase 4984
Calcium 4997
Chloride 3339
Creatine kinase 2346
Creatinine 4430
Free fatty acids 1280
Fructosamine 2150
Glucose 4974
Glycerol 1687
HDL-cholesterol 4116
Iron 3570
LDL-cholesterol 1877
Magnesium 1712
Phosphorus 4985
Potassium 3791
Sodium 3326
Total bilirubin 4390
Total cholesterol 4987
Total protein 4976
Triglyceride 1285
Triglycerides 3660
Urea 1287
Urea (Blood Urea Nitrogen - BUN) 3703length(unique(CC.stat$marker_symbol)) #3983[1] 3983length(unique(CC.stat$allele_symbol)) #4152[1] 4152length(unique(CC.stat$proc_param_name)) #27 # number of phenotypes in association statistics data set[1] 27length(unique(CC.data$proc_param_name)) #19 # number of phenotypes in final control data[1] 19pheno.list.stat <- unique(CC.stat$proc_param_name)
pheno.list.ctrl <- unique(CC.data$proc_param_name)
sum(pheno.list.stat %in% pheno.list.ctrl)[1] 19sum(pheno.list.ctrl %in% pheno.list.stat)[1] 19## extract common phenotype list
common.pheno.list <- sort(intersect(pheno.list.ctrl, pheno.list.stat))
common.pheno.list [1] "CC_Alanine aminotransferase" "CC_Albumin"
[3] "CC_Alkaline phosphatase" "CC_Alpha-amylase"
[5] "CC_Aspartate aminotransferase" "CC_Calcium"
[7] "CC_Chloride" "CC_Creatinine"
[9] "CC_Glucose" "CC_HDL-cholesterol"
[11] "CC_Iron" "CC_Phosphorus"
[13] "CC_Potassium" "CC_Sodium"
[15] "CC_Total bilirubin" "CC_Total cholesterol"
[17] "CC_Total protein" "CC_Triglycerides"
[19] "CC_Urea (Blood Urea Nitrogen - BUN)"length(common.pheno.list)[1] 19## Use summary statistics of common phenotypes
dim(CC.stat)[1] 95963 8CC.stat <- CC.stat %>% filter(proc_param_name %in% common.pheno.list)
dim(CC.stat)[1] 82339 8length(unique(CC.stat$proc_param_name))[1] 19Duplicates in gene-phenotype pair
mtest <- table(CC.stat$proc_param_name, CC.stat$marker_symbol)
mtest <-as.data.frame.matrix(mtest)
nmax <-max(mtest)
col_fun = colorRamp2(c(0, nmax), c("white", "red"))
col_fun(seq(0, nmax)) [1] "#FFFFFFFF" "#FFF1ECFF" "#FFE4DAFF" "#FFD6C8FF" "#FFC8B6FF" "#FFBAA4FF"
[7] "#FFAC93FF" "#FF9E81FF" "#FF8F70FF" "#FF805FFF" "#FF704FFF" "#FF5F3EFF"
[13] "#FF4B2DFF" "#FF331AFF" "#FF0000FF"ht = Heatmap(as.matrix(mtest), cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, col = col_fun,
row_names_gp = gpar(fontsize = 8), name="Count")`use_raster` is automatically set to TRUE for a matrix with more than
2000 columns You can control `use_raster` argument by explicitly
setting TRUE/FALSE to it.
Set `ht_opt$message = FALSE` to turn off this message.'magick' package is suggested to install to give better rasterization.
Set `ht_opt$message = FALSE` to turn off this message.draw(ht)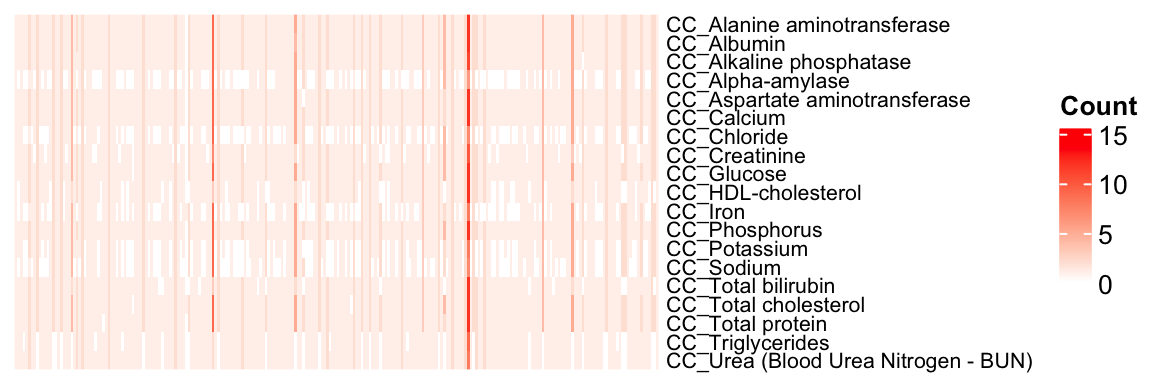
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Using Stouffer’s method, merge multiple z-scores of a gene-phenotype pair into a z-score
## sum(z-score)/sqrt(# of zscore)
sumz <- function(z){ sum(z)/sqrt(length(z)) }
CC.z = CC.stat %>%
dplyr::select(marker_symbol, proc_param_name, z_score) %>%
na.omit() %>%
group_by(marker_symbol, proc_param_name) %>%
summarize(zscore = sumz(z_score)) ## combine z-scores`summarise()` has grouped output by 'marker_symbol'. You can override using the
`.groups` argument.dim(CC.z)[1] 58770 3Make z-score matrix (long to wide)
nan2na <- function(df){
out <- data.frame(sapply(df, function(x) ifelse(is.nan(x), NA, x)))
colnames(out) <- colnames(df)
out
}
CC.zmat = dcast(CC.z, marker_symbol ~ proc_param_name, value.var = "zscore",
fun.aggregate = mean) %>% tibble::column_to_rownames(var="marker_symbol")
CC.zmat = nan2na(CC.zmat) #convert nan to na
dim(CC.zmat)[1] 3983 19dim(CC.zmat)[1] 3983 19saveRDS(CC.zmat, file = "data/CC.zmat.rds")
id.mat <- 1*(!is.na(CC.zmat)) # multiply 1 to make this matrix numeric
nrow(as.data.frame(colSums(id.mat)))[1] 19dim(id.mat)[1] 3983 19## heatmap of gene - phenotype (red: tested, white: untested)
ht = Heatmap(t(id.mat),
cluster_rows = T, clustering_distance_rows ="binary",
cluster_columns = T, clustering_distance_columns = "binary",
show_row_dend = F, show_column_dend = F, # do not show dendrogram
show_column_names = F, col = c("white","red"),
row_names_gp = gpar(fontsize = 10), name="Missing")`use_raster` is automatically set to TRUE for a matrix with more than
2000 columns You can control `use_raster` argument by explicitly
setting TRUE/FALSE to it.
Set `ht_opt$message = FALSE` to turn off this message.'magick' package is suggested to install to give better rasterization.
Set `ht_opt$message = FALSE` to turn off this message.draw(ht)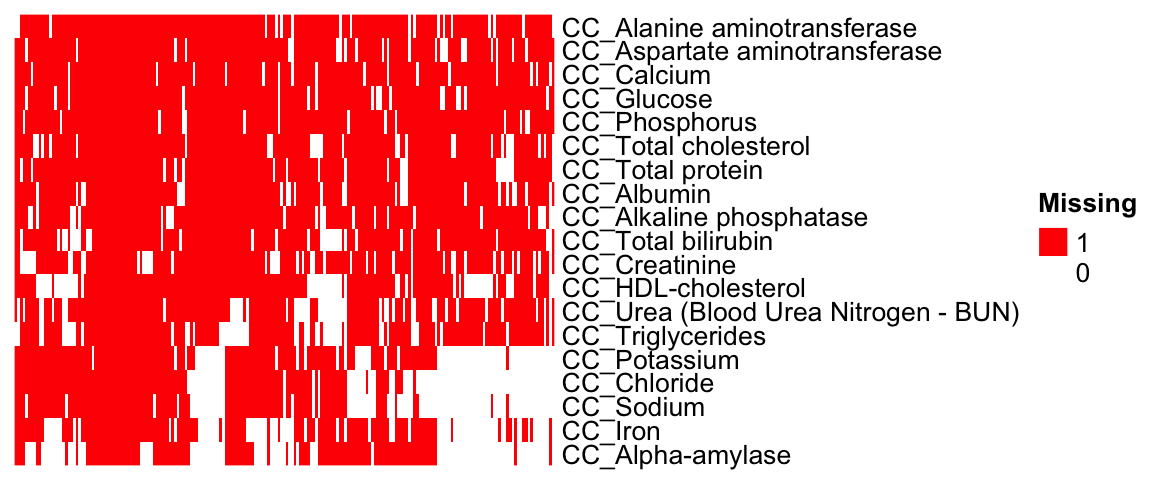
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Z-score Distribution
We plot association Z-score distribution for each phenotype.
ggplot(melt(CC.zmat), aes(x=value)) +
geom_histogram() +
facet_wrap(~variable, scales="free", ncol=5)+
theme(strip.text.x = element_text(size = 6))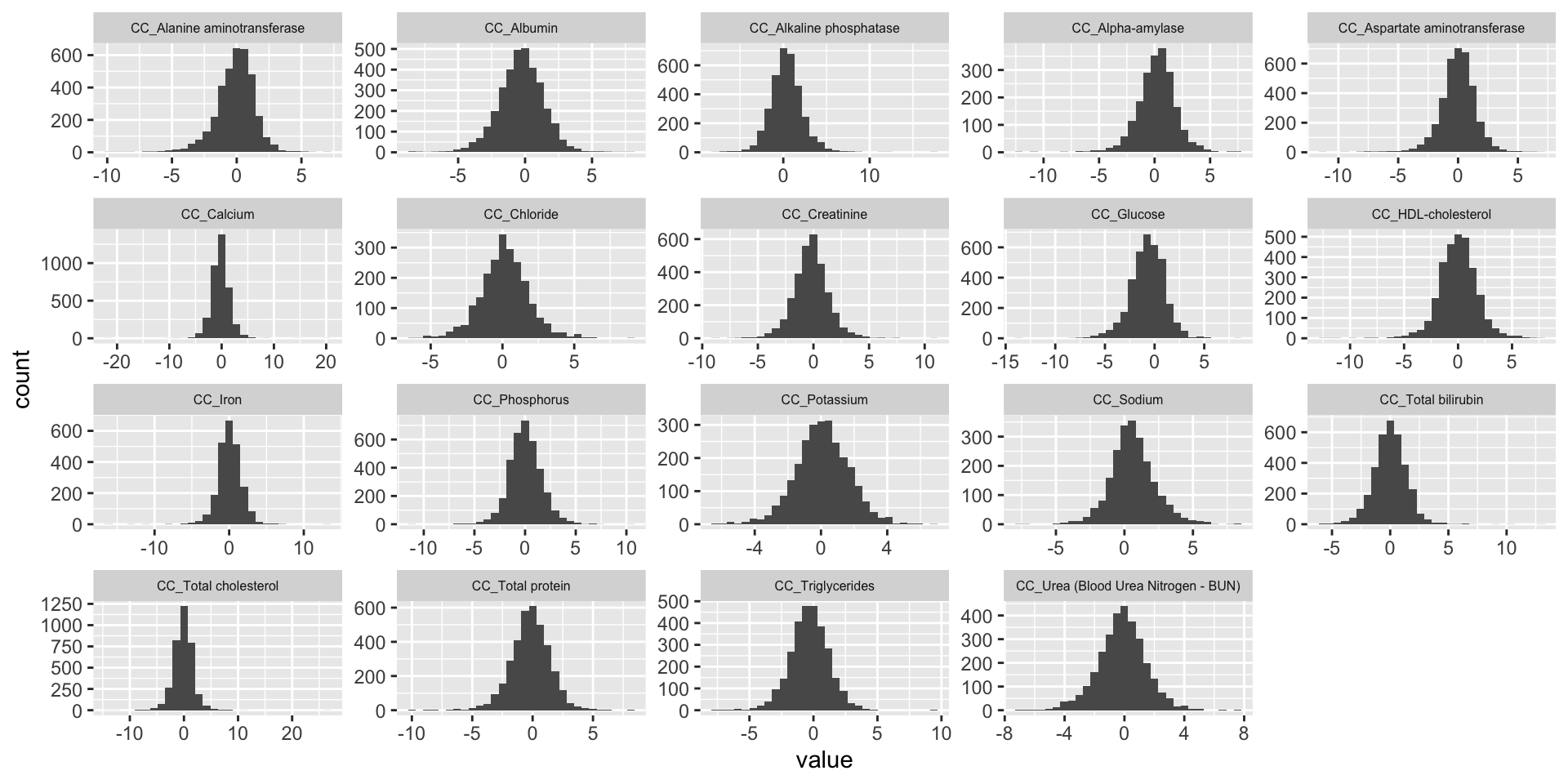
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Estimate genetic correlation matrix between phenotypes using Zscores
Here, we estimate the genetic correlations between phenotypes using association Z-score matrix (num of genes:3983, num of phenotypes 19).
CC.zmat <- CC.zmat[,common.pheno.list]
CC.zcor = cor(CC.zmat, use="pairwise.complete.obs")
ht = Heatmap(CC.zcor, cluster_rows = T, cluster_columns = T, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 10),
name="Genetic Corr (Z-score)"
)
draw(ht)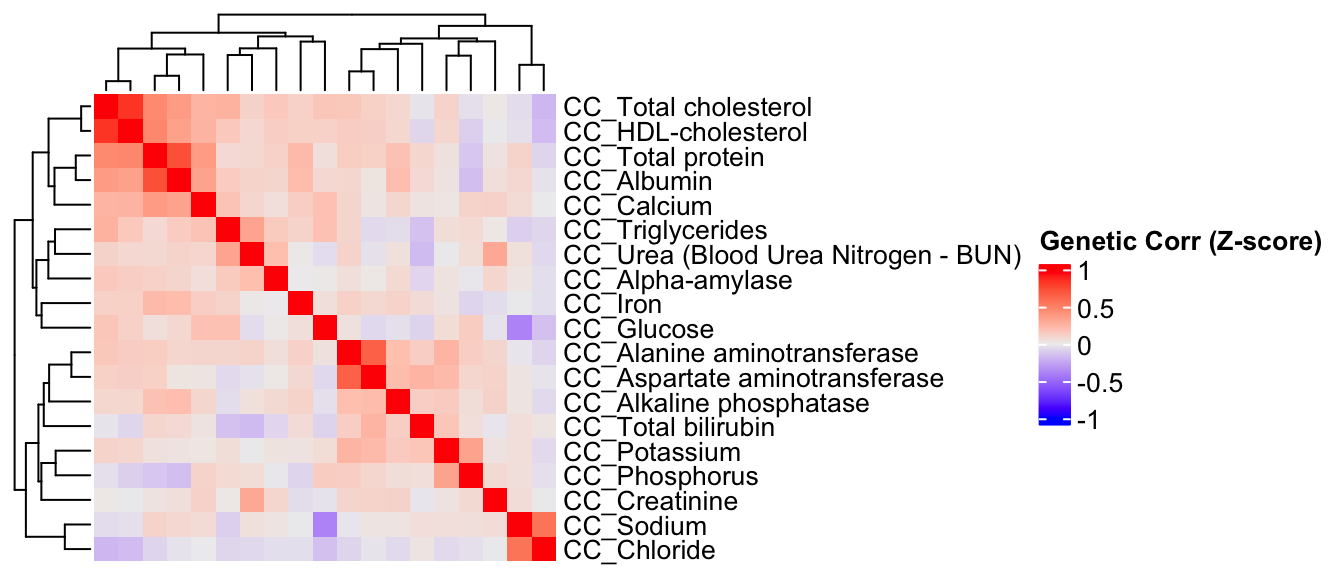
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
Phenotype Corr VS Genetic Corr btw phenotypes
We compare a correlation matrix obtained using control mice phenotype data v.s. a genetic correlation matrix estimated using association Z-scores. As you can see, both correlation heatmaps have similar correlation pattern.
CC.cor.rank.fig <- CC.cor.rank[common.pheno.list,common.pheno.list]
CC.cor.fig <- CC.cor[common.pheno.list,common.pheno.list]
CC.cor.combat.fig <- CC.cor.combat[common.pheno.list, common.pheno.list]
CC.zcor.fig <- CC.zcor
ht = Heatmap(CC.cor.rank.fig, cluster_rows = TRUE, cluster_columns = TRUE, show_column_names = F, #col = col_fun,
show_row_dend = F, show_column_dend = F, # do not show dendrogram
row_names_gp = gpar(fontsize = 8), column_title="Phenotype Corr (RankZ, Pearson)", column_title_gp = gpar(fontsize = 8),
name="Corr")
pheno.order <- row_order(ht)Warning: The heatmap has not been initialized. You might have different results
if you repeatedly execute this function, e.g. when row_km/column_km was
set. It is more suggested to do as `ht = draw(ht); row_order(ht)`.#draw(ht)
CC.cor.rank.fig <- CC.cor.rank.fig[pheno.order,pheno.order]
ht1 = Heatmap(CC.cor.rank.fig, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
show_row_dend = F, show_column_dend = F, # do not show dendrogram
row_names_gp = gpar(fontsize = 8), column_title="Phenotype Corr (RankZ, Pearson)", column_title_gp = gpar(fontsize = 8),
name="Corr")
CC.cor.fig <- CC.cor.fig[pheno.order,pheno.order]
ht2 = Heatmap(CC.cor.fig, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 8), column_title="Phenotype Corr (Spearman)", column_title_gp = gpar(fontsize = 8),
name="Corr")
CC.cor.combat.fig <- CC.cor.combat.fig[pheno.order,pheno.order]
ht3 = Heatmap(CC.cor.combat.fig, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 8), column_title="Phenotype Corr (Combat, Pearson)", column_title_gp = gpar(fontsize = 8),
name="Corr")
CC.zcor.fig <- CC.zcor.fig[pheno.order,pheno.order]
ht4 = Heatmap(CC.zcor.fig, cluster_rows = FALSE, cluster_columns = FALSE, show_column_names = F, #col = col_fun,
row_names_gp = gpar(fontsize = 8), column_title="Genetic Corr (Pearson)", column_title_gp = gpar(fontsize = 8),
name="Corr"
)
draw(ht1+ht2+ht3+ht4)Warning: Heatmap/annotation names are duplicated: CorrWarning: Heatmap/annotation names are duplicated: Corr, CorrWarning: Heatmap/annotation names are duplicated: Corr, Corr, Corr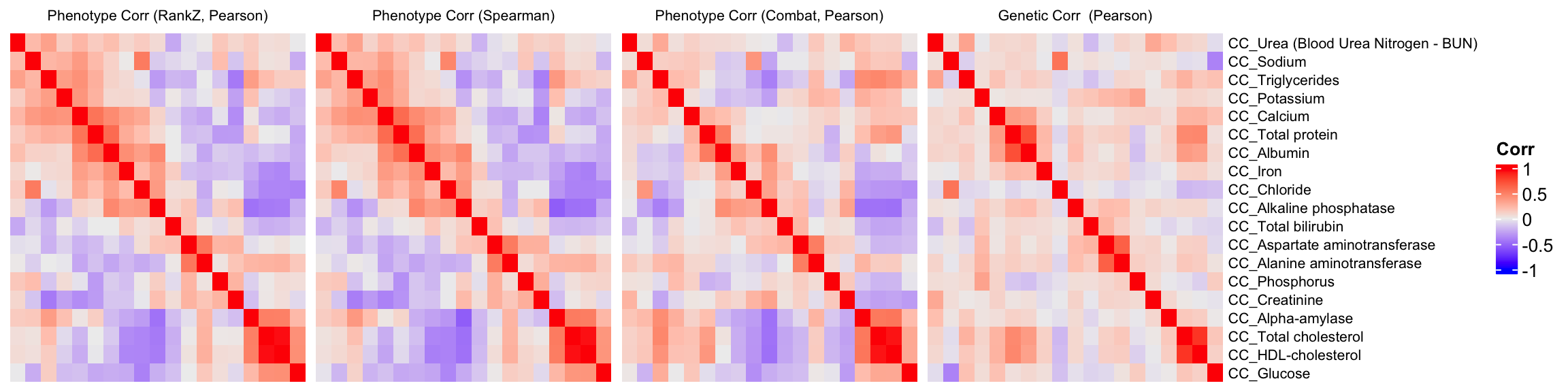
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
png(file="docs/figure/figures.Rmd/cors_CC.png", width=800, height=250)
draw(ht1+ht2+ht3+ht4)Warning: Heatmap/annotation names are duplicated: CorrWarning: Heatmap/annotation names are duplicated: Corr, CorrWarning: Heatmap/annotation names are duplicated: Corr, Corr, Corrdev.off()quartz_off_screen
2 Test of the correlation between genetic correlation matrices
We use the Mantel’s test for testing the correlation between two distance matrices.
####################
# Use Mantel test
# https://stats.idre.ucla.edu/r/faq/how-can-i-perform-a-mantel-test-in-r/
# install.packages("ade4")
library(ade4)
to.upper<-function(X) X[upper.tri(X,diag=FALSE)]
a1 <- to.upper(CC.cor.fig)
a2 <- to.upper(CC.cor.rank.fig)
a3 <- to.upper(CC.cor.combat.fig)
a4 <- to.upper(CC.zcor.fig)
plot(a4, a1)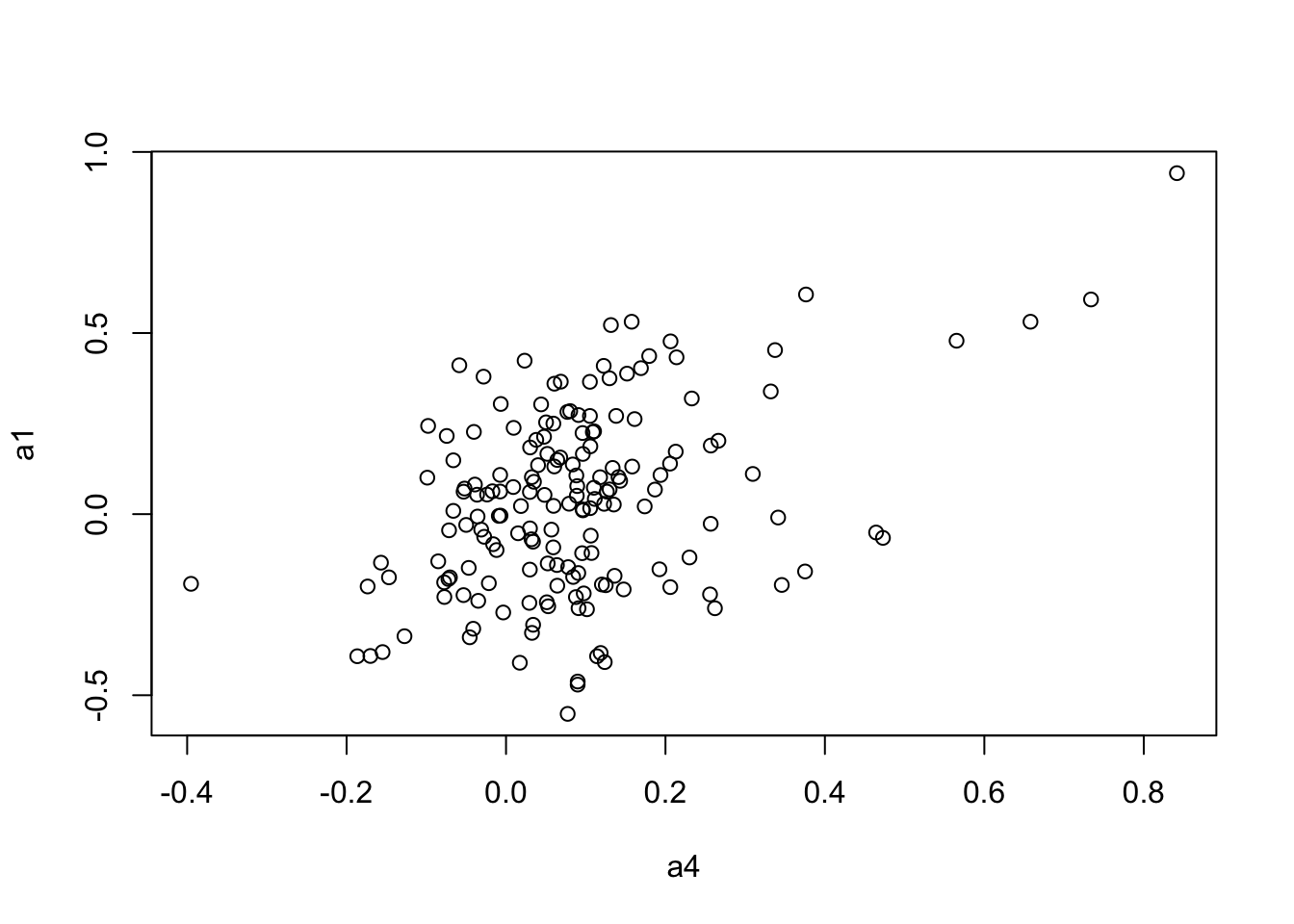
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
plot(a4, a2)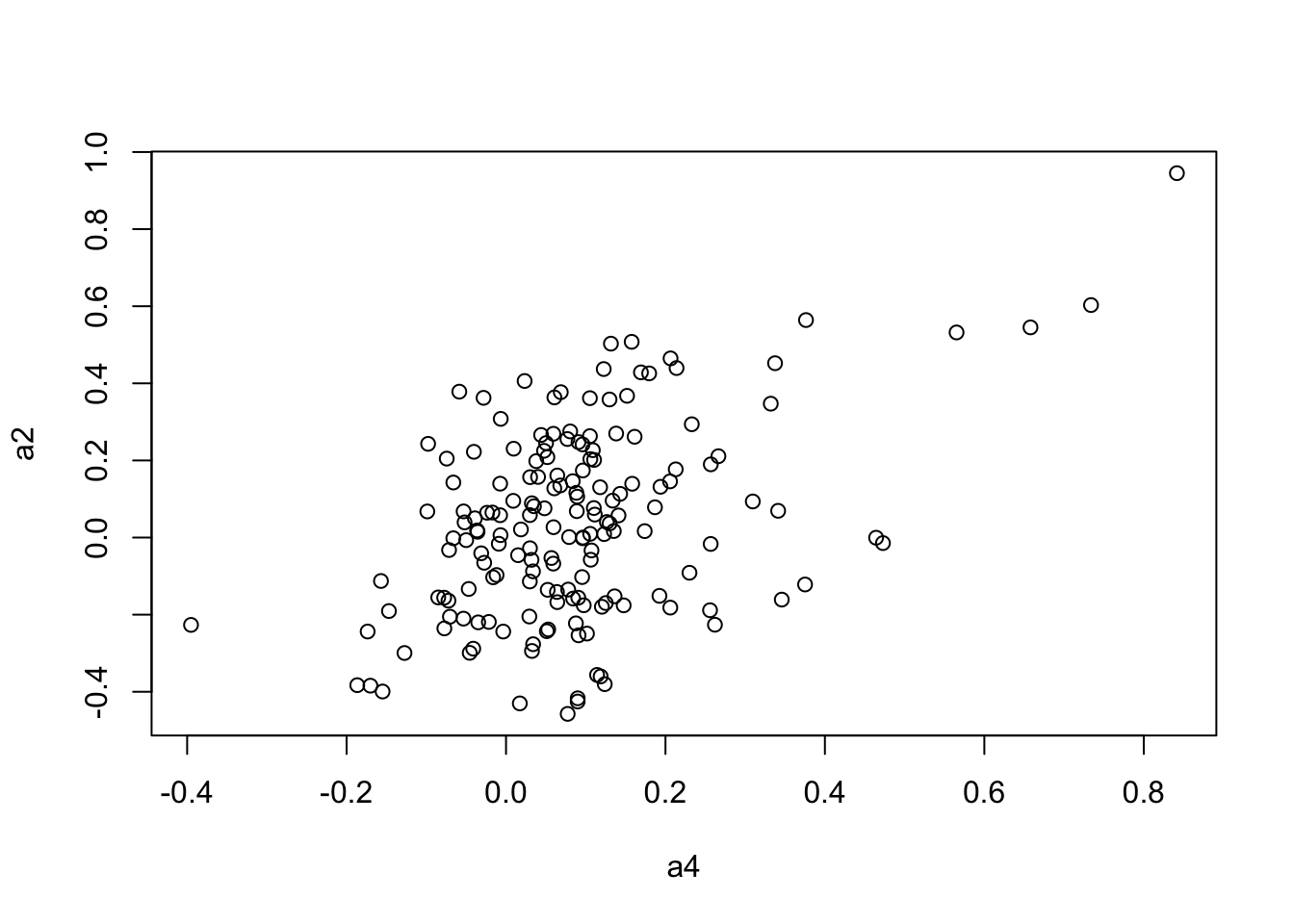
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
plot(a4, a3)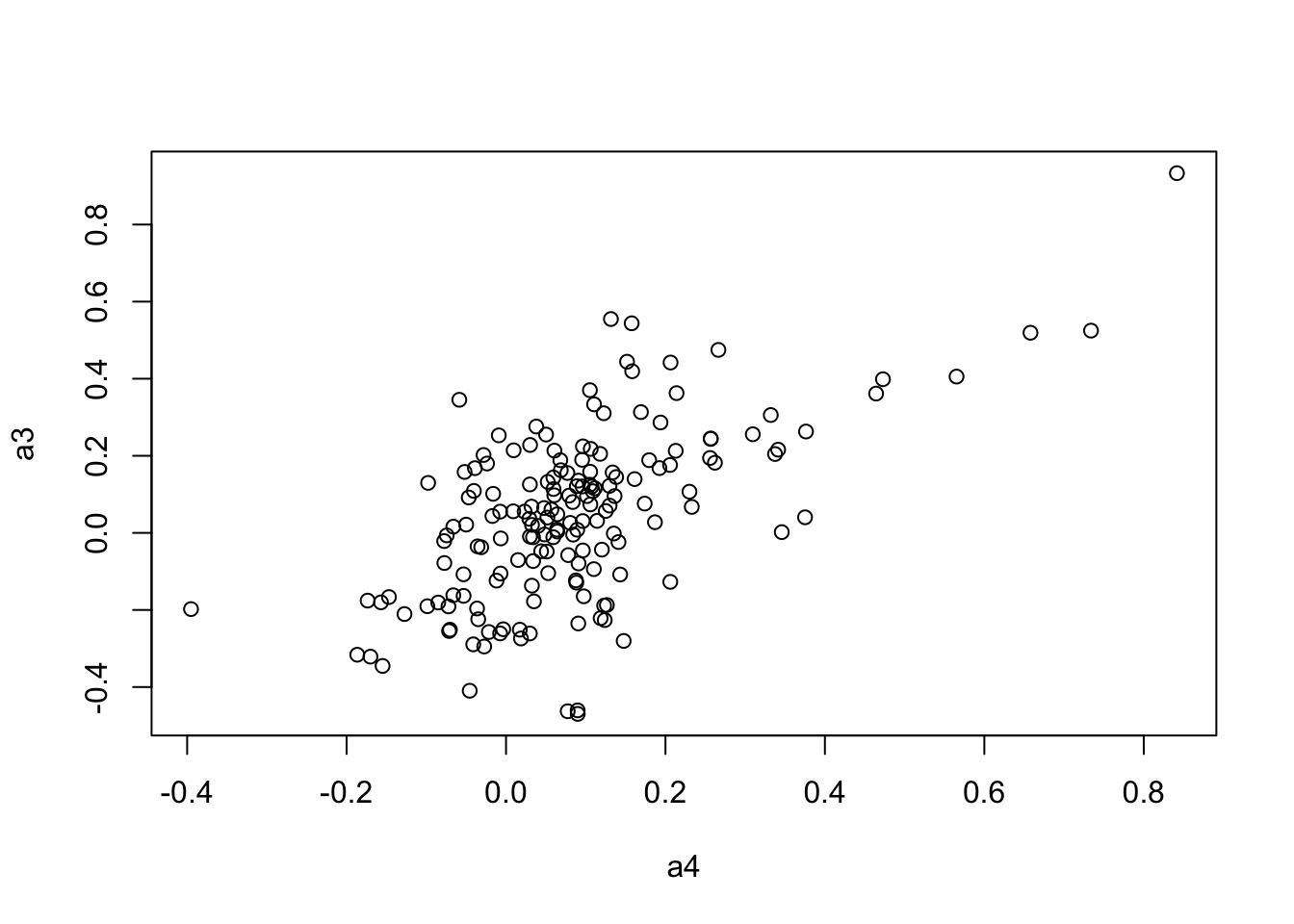
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
mantel.rtest(as.dist(1-CC.cor.fig), as.dist(1-CC.zcor.fig), nrepet = 9999) #nrepet = number of permutationsMonte-Carlo test
Call: mantelnoneuclid(m1 = m1, m2 = m2, nrepet = nrepet)
Observation: 0.4269902
Based on 9999 replicates
Simulated p-value: 1e-04
Alternative hypothesis: greater
Std.Obs Expectation Variance
5.688672800 0.001308730 0.005599478 mantel.rtest(as.dist(1-CC.cor.rank.fig), as.dist(1-CC.zcor.fig), nrepet = 9999)Monte-Carlo test
Call: mantelnoneuclid(m1 = m1, m2 = m2, nrepet = nrepet)
Observation: 0.4639696
Based on 9999 replicates
Simulated p-value: 1e-04
Alternative hypothesis: greater
Std.Obs Expectation Variance
6.125340224 0.001131372 0.005709499 mantel.rtest(as.dist(1-CC.cor.combat.fig), as.dist(1-CC.zcor.fig), nrepet = 9999)Monte-Carlo test
Call: mantelnoneuclid(m1 = m1, m2 = m2, nrepet = nrepet)
Observation: 0.6175711
Based on 9999 replicates
Simulated p-value: 1e-04
Alternative hypothesis: greater
Std.Obs Expectation Variance
8.301808421 -0.001605264 0.005562671 Test KOMPUTE imputation algorithm
Load KOMPUTE package
if(!"kompute" %in% rownames(installed.packages())){
library(devtools)
devtools::install_github("dleelab/kompute")
}
library(kompute)Simulation study - imputed vs measured
We randomly select measured gene-phenotype association z-scores, mask those, impute them using KOMPUTE method. Then we compare the imputed z-scores to the measured ones.
zmat <-t(CC.zmat)
dim(zmat)[1] 19 3983## filter genes with na < 10
zmat0 <- is.na(zmat)
num.na<-colSums(zmat0)
summary(num.na) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.000 2.000 4.000 4.245 6.000 17.000 zmat <- zmat[,num.na<10]
dim(zmat)[1] 19 3851#pheno.cor <- CC.cor.fig
#pheno.cor <- CC.cor.rank.fig
pheno.cor <- CC.cor.combat.fig
#pheno.cor <- CC.zcor.fig
zmat <- zmat[rownames(pheno.cor),,drop=FALSE]
rownames(zmat) [1] "CC_Urea (Blood Urea Nitrogen - BUN)" "CC_Sodium"
[3] "CC_Triglycerides" "CC_Potassium"
[5] "CC_Calcium" "CC_Total protein"
[7] "CC_Albumin" "CC_Iron"
[9] "CC_Chloride" "CC_Alkaline phosphatase"
[11] "CC_Total bilirubin" "CC_Aspartate aminotransferase"
[13] "CC_Alanine aminotransferase" "CC_Phosphorus"
[15] "CC_Creatinine" "CC_Alpha-amylase"
[17] "CC_Total cholesterol" "CC_HDL-cholesterol"
[19] "CC_Glucose" rownames(pheno.cor) [1] "CC_Urea (Blood Urea Nitrogen - BUN)" "CC_Sodium"
[3] "CC_Triglycerides" "CC_Potassium"
[5] "CC_Calcium" "CC_Total protein"
[7] "CC_Albumin" "CC_Iron"
[9] "CC_Chloride" "CC_Alkaline phosphatase"
[11] "CC_Total bilirubin" "CC_Aspartate aminotransferase"
[13] "CC_Alanine aminotransferase" "CC_Phosphorus"
[15] "CC_Creatinine" "CC_Alpha-amylase"
[17] "CC_Total cholesterol" "CC_HDL-cholesterol"
[19] "CC_Glucose" colnames(pheno.cor) [1] "CC_Urea (Blood Urea Nitrogen - BUN)" "CC_Sodium"
[3] "CC_Triglycerides" "CC_Potassium"
[5] "CC_Calcium" "CC_Total protein"
[7] "CC_Albumin" "CC_Iron"
[9] "CC_Chloride" "CC_Alkaline phosphatase"
[11] "CC_Total bilirubin" "CC_Aspartate aminotransferase"
[13] "CC_Alanine aminotransferase" "CC_Phosphorus"
[15] "CC_Creatinine" "CC_Alpha-amylase"
[17] "CC_Total cholesterol" "CC_HDL-cholesterol"
[19] "CC_Glucose" npheno <- nrow(zmat)
## percentage of missing Z-scores in the original data
100*sum(is.na(zmat))/(nrow(zmat)*ncol(zmat)) # 21%[1] 21.11823nimp <- 2000 # # of missing/imputed Z-scores
set.seed(1111)
## find index of all measured zscores
all.i <- 1:(nrow(zmat)*ncol(zmat))
measured <- as.vector(!is.na(as.matrix(zmat)))
measured.i <- all.i[measured]
## mask 2000 measured z-scores
mask.i <- sort(sample(measured.i, nimp))
org.z = as.matrix(zmat)[mask.i]
zvec <- as.vector(as.matrix(zmat))
zvec[mask.i] <- NA
zmat.imp <- matrix(zvec, nrow=npheno)
rownames(zmat.imp) <- rownames(zmat)Run KOMPUTE method
kompute.res <- kompute(zmat.imp, pheno.cor, 0.01)
KOMPute running...# of genes: 3851# of phenotypes: 19# of imputed Z-scores: 17452# measured vs imputed
length(org.z)[1] 2000imp.z <- as.matrix(kompute.res$zmat)[mask.i]
imp.info <- as.matrix(kompute.res$infomat)[mask.i]
plot(imp.z, org.z)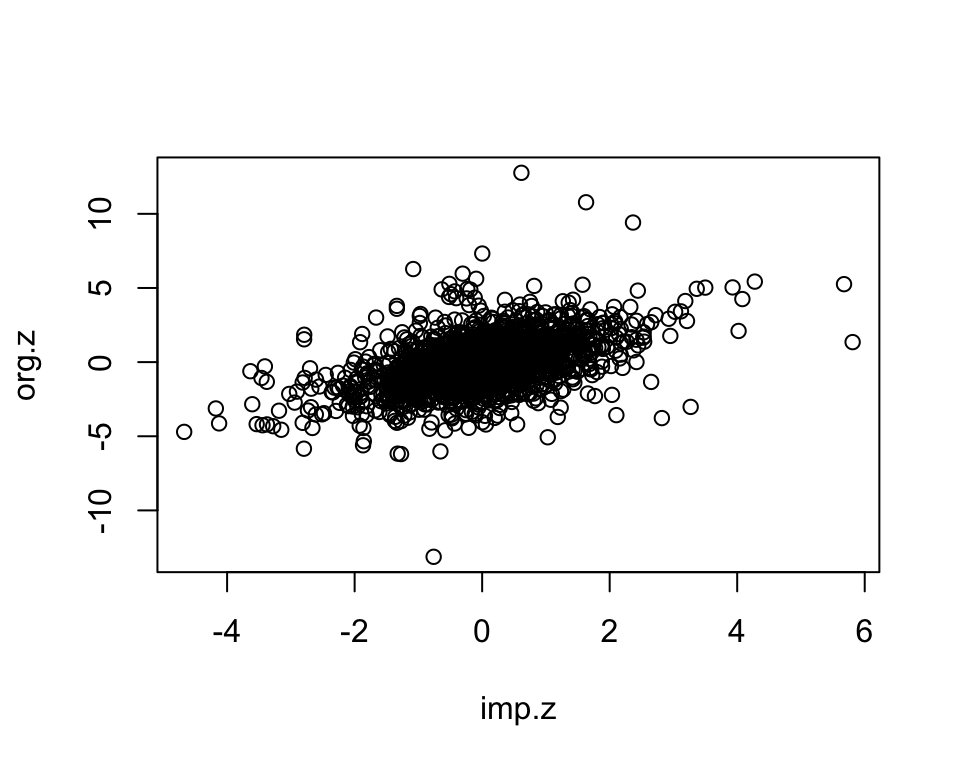
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
imp <- data.frame(org.z=org.z, imp.z=imp.z, info=imp.info)
dim(imp)[1] 2000 3imp <- imp[complete.cases(imp),]
imp <- subset(imp, info>=0 & info <= 1)
dim(imp)[1] 2000 3cor.val <- round(cor(imp$imp.z, imp$org.z), digits=3)
cor.val[1] 0.477plot(imp$imp.z, imp$org.z)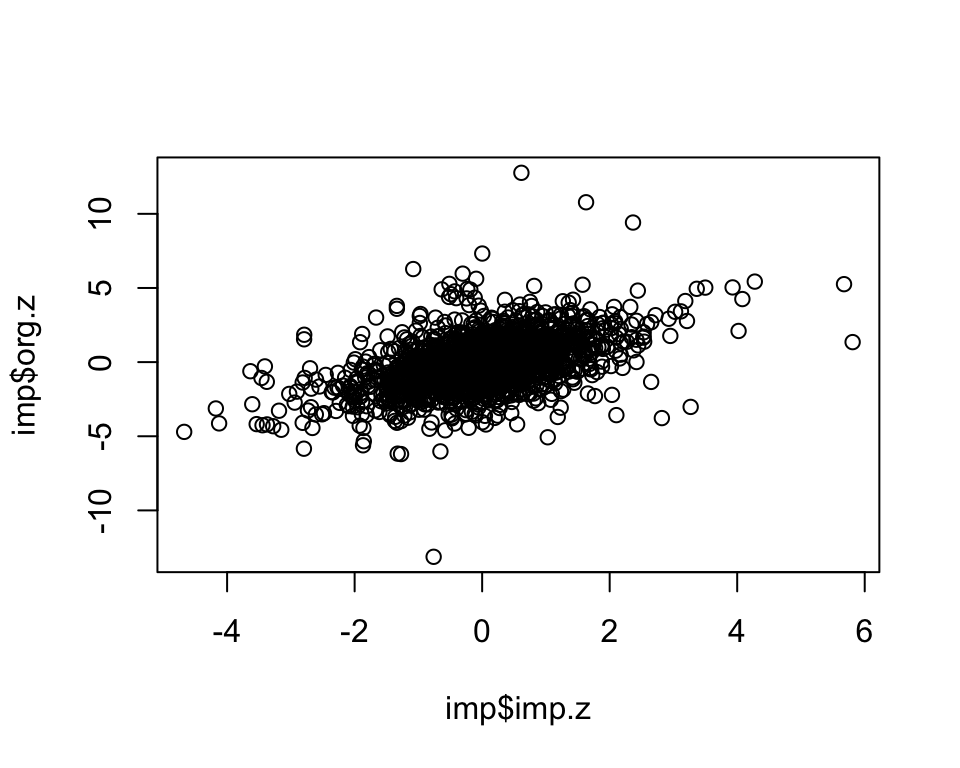
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
info.cutoff <- 0.8
imp.sub <- subset(imp, info>info.cutoff)
dim(imp.sub)[1] 197 3summary(imp.sub$imp.z) Min. 1st Qu. Median Mean 3rd Qu. Max.
-4.67331 -0.93724 0.03962 0.01791 1.04483 4.27590 summary(imp.sub$info) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.8682 0.8760 0.8783 0.8773 0.8796 0.8804 cor.val <- round(cor(imp.sub$imp.z, imp.sub$org.z), digits=3)
cor.val[1] 0.846g <- ggplot(imp.sub, aes(x=imp.z, y=org.z, col=info)) +
geom_point() +
labs(title=paste0("IMPC Behavior Data (CC), Info>", info.cutoff, ", Cor=",cor.val),
x="Imputed Z-scores", y = "Measured Z-scores", col="Info") +
theme_minimal()
g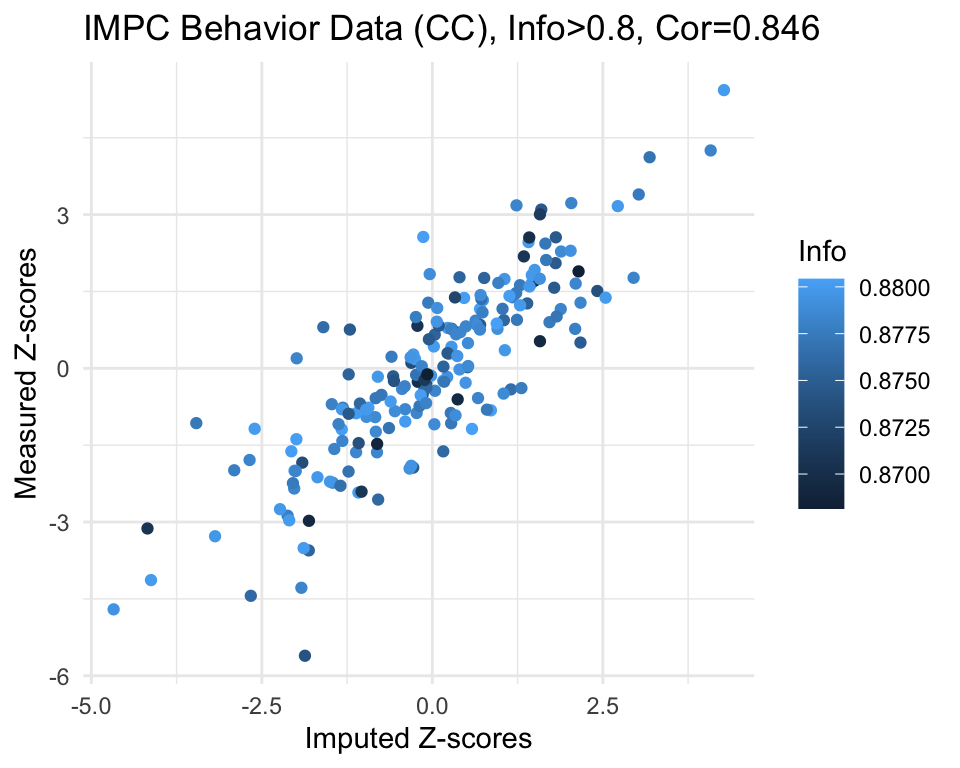
| Version | Author | Date |
|---|---|---|
| 7685a09 | statsleelab | 2023-01-10 |
# save plot
png(file="docs/figure/figures.Rmd/sim_results_CC.png", width=600, height=350)
g
dev.off()quartz_off_screen
2
sessionInfo()R version 4.2.1 (2022-06-23)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.7
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] kompute_0.1.0 ade4_1.7-20 sva_3.44.0
[4] BiocParallel_1.30.3 genefilter_1.78.0 mgcv_1.8-40
[7] nlme_3.1-158 lme4_1.1-31 Matrix_1.5-1
[10] RNOmni_1.0.1 ComplexHeatmap_2.12.1 circlize_0.4.15
[13] RColorBrewer_1.1-3 tidyr_1.2.0 ggplot2_3.4.1
[16] reshape2_1.4.4 dplyr_1.0.9 data.table_1.14.2
[19] workflowr_1.7.0.1
loaded via a namespace (and not attached):
[1] minqa_1.2.5 colorspace_2.1-0 rjson_0.2.21
[4] rprojroot_2.0.3 XVector_0.36.0 GlobalOptions_0.1.2
[7] fs_1.5.2 clue_0.3-62 rstudioapi_0.13
[10] farver_2.1.1 bit64_4.0.5 AnnotationDbi_1.58.0
[13] fansi_1.0.4 codetools_0.2-18 splines_4.2.1
[16] doParallel_1.0.17 cachem_1.0.6 knitr_1.39
[19] jsonlite_1.8.0 nloptr_2.0.3 annotate_1.74.0
[22] cluster_2.1.3 png_0.1-8 compiler_4.2.1
[25] httr_1.4.3 assertthat_0.2.1 fastmap_1.1.0
[28] limma_3.52.4 cli_3.6.0 later_1.3.0
[31] htmltools_0.5.3 tools_4.2.1 gtable_0.3.1
[34] glue_1.6.2 GenomeInfoDbData_1.2.8 Rcpp_1.0.10
[37] Biobase_2.56.0 jquerylib_0.1.4 vctrs_0.5.2
[40] Biostrings_2.64.0 iterators_1.0.14 xfun_0.31
[43] stringr_1.4.0 ps_1.7.1 lifecycle_1.0.3
[46] XML_3.99-0.10 edgeR_3.38.4 getPass_0.2-2
[49] MASS_7.3-58.1 zlibbioc_1.42.0 scales_1.2.1
[52] promises_1.2.0.1 parallel_4.2.1 yaml_2.3.5
[55] memoise_2.0.1 sass_0.4.2 stringi_1.7.8
[58] RSQLite_2.2.15 highr_0.9 S4Vectors_0.34.0
[61] foreach_1.5.2 BiocGenerics_0.42.0 boot_1.3-28
[64] shape_1.4.6 GenomeInfoDb_1.32.3 rlang_1.0.6
[67] pkgconfig_2.0.3 matrixStats_0.62.0 bitops_1.0-7
[70] evaluate_0.16 lattice_0.20-45 purrr_0.3.4
[73] labeling_0.4.2 bit_4.0.4 processx_3.7.0
[76] tidyselect_1.2.0 plyr_1.8.7 magrittr_2.0.3
[79] R6_2.5.1 IRanges_2.30.0 generics_0.1.3
[82] DBI_1.1.3 pillar_1.8.1 whisker_0.4
[85] withr_2.5.0 survival_3.3-1 KEGGREST_1.36.3
[88] RCurl_1.98-1.8 tibble_3.1.8 crayon_1.5.1
[91] utf8_1.2.3 rmarkdown_2.14 GetoptLong_1.0.5
[94] locfit_1.5-9.6 blob_1.2.3 callr_3.7.1
[97] git2r_0.30.1 digest_0.6.29 xtable_1.8-4
[100] httpuv_1.6.5 stats4_4.2.1 munsell_0.5.0
[103] bslib_0.4.0